Perturb-seq - Perturb-seq
Perturb-seq (также известный как CRISP-seq и CROP-seq) относится к высокопроизводительному методу выполнения секвенирование одноклеточной РНК (scRNA-seq) на объединенных экранах генетических нарушений.[1][2][3] Perturb-seq объединяет мультиплексированные CRISPR опосредованная инактивация генов с секвенированием одноклеточной РНК для оценки всестороннего экспрессия гена фенотипы для каждого возмущения. Вывод функции гена путем применения генетических возмущений к сбить или же нокаутировать ген и изучение результирующего фенотипа известно как обратная генетика. Perturb-seq - это подход обратной генетики, который позволяет исследовать фенотипы на уровне транскриптом, чтобы выяснить функции генов во многих клетках массовым образом.
Протокол Perturb-seq использует CRISPR технология для инактивации определенных генов и Штрих-кодирование ДНК каждой направляющей РНК, чтобы все возмущения можно было объединить вместе, а затем деконволютировать с отнесением каждого фенотипа к определенному направляющая РНК.[1][2] На основе капель микрофлюидика платформы (или другие методы сортировки и разделения клеток) используются для выделения отдельных клеток, а затем выполняется scRNA-seq для генерации экспрессия гена профили для каждой ячейки. По завершении протокола, биоинформатика проводятся анализы, чтобы связать каждую конкретную клетку и нарушение с транскриптомным профилем, который характеризует последствия инактивации каждого гена.
В декабрьском выпуске журнала Клетка В журнале были опубликованы две сопутствующие статьи, в каждой из которых была представлена и описана эта техника.[1][2] Третий документ, описывающий концептуально подобный подход (названный CRISP-seq), также был опубликован в том же номере.[4] В октябре 2016 года метод CROP-seq для одноклеточного CRISPR-скрининга был представлен в препринте на bioRxiv[5] а позже опубликовано в Природные методы журнал.[3] В то время как в каждой статье были изложены основные принципы сочетания опосредованного CRISPR возмущения с scRNA-seq, их экспериментальные, технологические и аналитические подходы различались по нескольким аспектам, чтобы исследовать отдельные биологические вопросы, демонстрируя широкую полезность этой методологии. Например, статья CRISPR-seq продемонстрировала возможность in vivo исследования с использованием этой технологии, а протокол CROP-seq упрощает использование больших экранов, предоставляя вектор, который делает саму направляющую РНК читаемой (вместо того, чтобы полагаться на выраженные штрих-коды), что позволяет выполнять одноэтапное клонирование направляющей РНК.[6]
Экспериментальный рабочий процесс
Разработка и выбор библиотеки РНК CRISPR Single Guide
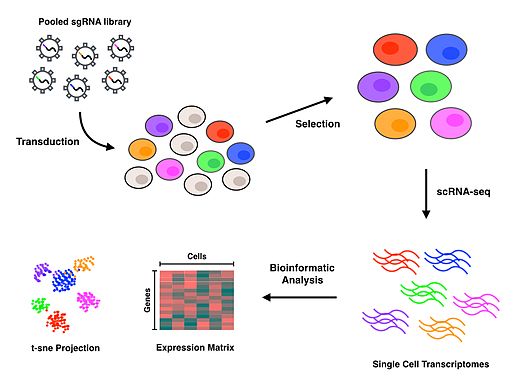
Объединенный CRISPR библиотеки инактивация генов может происходить в форме нокаута или интерференции. Нокаут-библиотеки нарушают гены из-за двухцепочечных разрывов, которые приводят к возникновению ошибок негомологичное соединение концов путь восстановления для введения деструктивных вставок или делеций. CRISPR вмешательство (CRISPRi) с другой стороны, использует каталитически неактивный нуклеаза физически заблокировать РНК-полимераза, эффективно предотвращая или останавливая транскрипция.[7] Perturb-seq использовался как с нокаутом, так и с CRISPRi в Dixit et al. бумага [2] и Adamson et al. бумага,[1] соответственно.
Объединение всех направляющих РНК в один экран зависит от штрих-кодов ДНК, которые действуют как идентификаторы для каждой уникальной направляющей РНК. Существует несколько коммерчески доступных объединенных библиотек CRISPR, включая библиотеку направляющих штрих-кодов, использованную в исследовании Adamson et al.[1] Библиотеки CRISPR также могут быть созданы с использованием инструментов для проектирования sgRNA, многие из которых перечислены на Инструменты CRISPR / cas9 Страница Википедии.
Лентивирусные векторы
Дизайн вектора экспрессии sgRNA будет во многом зависеть от проведенного эксперимента, но требует следующих центральных компонентов:
- Промоутер
- Сайты ограничения
- Грунтовка Участок связывания
- sgRNA
- Направляющий штрих-код
- Репортерный ген:
- Флуоресцентный ген: Векторы часто конструируются так, чтобы включать ген, кодирующий флуоресцентный белок, так что успешно трансдуцированные клетки можно визуально и количественно оценить по их экспрессии.
- Устойчивость к антибиотикам ген: Подобно флуоресцентным маркерам, гены устойчивости к антибиотикам часто включаются в векторы, чтобы обеспечить селекцию успешно трансдуцированных клеток.
- CRISPR-ассоциированная эндонуклеаза: Cas9 или другие CRISPR-ассоциированные эндонуклеазы, такие как Cpf1 должны быть введены в клетки, которые не экспрессируют их эндогенно. Из-за большого размера этих генов можно использовать двухвекторную систему для экспрессии эндонуклеазы отдельно от вектора экспрессии sgRNA.[8]
Трансдукция и отбор
Клетки обычно преобразованный с Множественность инфекции (MOI) от 0,4 до 0,6 лентивирусные частицы на клетку, чтобы максимизировать вероятность получения наибольшего количества клеток, содержащих одну направляющую РНК.[8][9] Если представляют интерес эффекты одновременных возмущений, можно применить более высокий MOI для увеличения количества трансдуцированных клеток с более чем одной направляющей РНК. Затем выполняется отбор успешно трансдуцированных клеток с использованием флуоресцентного анализа или анализа антибиотиков, в зависимости от репортерного гена, используемого в векторе экспрессии.
Подготовка одноклеточной библиотеки
После того, как были отобраны успешно трансдуцированные клетки, необходимо выделить отдельные клетки для проведения scRNA-seq. Perturb-seq и CROP-seq были выполнены с использованием капельной технологии для выделения отдельных клеток,[1][2][3] в то время как тесно связанный CRISP-seq был выполнен с использованием подхода на основе микролунок.[4] После того, как клетки были изолированы на уровне отдельной клетки, обратная транскрипция происходит амплификация и секвенирование для получения профилей экспрессии генов для каждой клетки. Многие подходы scRNA-seq включают уникальные молекулярные идентификаторы (UMI) и штрих-коды клеток на этапе обратной транскрипции для индексации отдельных молекул РНК и клеток соответственно. Эти дополнительные штрих-коды служат для количественной оценки транскриптов РНК и для связывания каждой из последовательностей с их клеткой происхождения.
Биоинформатический анализ
Выравнивание и обработка считывания выполняются для обеспечения качества считывания эталонного генома. Деконволюция клеточных штрих-кодов, направляющих штрих-кодов и UMI позволяет связывать направляющие РНК с клетками, которые их содержат, тем самым позволяя профилю экспрессии генов каждой клетки быть связанным с определенным возмущением. Дальнейшие последующие анализы транскрипционных профилей будут полностью зависеть от интересующего биологического вопроса. T-распределенное стохастическое соседнее вложение (t-SNE) обычно используется машинное обучение алгоритм для визуализации многомерных данных, полученных в результате scRNA-seq, на 2-мерной диаграмме рассеяния.[1][4][10] Авторы, впервые выполнившие Perturb-seq, разработали внутреннюю вычислительную структуру под названием MIMOSCA, которая прогнозирует эффекты каждого возмущения с использованием линейной модели и доступна в открытом репозитории программного обеспечения.[11]
Преимущества и ограничения
Perturb-seq использует современные технологии молекулярной биологии для интеграции многоэтапного рабочего процесса, который сочетает высокопроизводительный скрининг со сложными фенотипическими результатами. По сравнению с альтернативными методами, используемыми для нокдауна или нокаута генов, такими как РНКи, нуклеазы цинковых пальцев или же эффекторные нуклеазы, подобные активаторам транскрипции (TALEN), применение отклонений на основе CRISPR обеспечивает большую конкретность, эффективность и простоту использования.[8][12] Еще одно преимущество этого протокола заключается в том, что в то время как большинство подходов к скринингу могут анализировать только простые фенотипы, такие как жизнеспособность клеток, scRNA-seq позволяет получить гораздо более богатое фенотипическое считывание с количественными измерениями экспрессии генов во многих клетках одновременно.
Однако, несмотря на то, что большой и полный объем данных может быть преимуществом, он также может представлять серьезную проблему. Известно, что считывание экспрессии РНК одной клетки дает «шумные» данные со значительным количеством ложных срабатываний.[13] И большой размер, и шум, связанный с scRNA-seq, вероятно, потребуют новых и мощных вычислительных методов и конвейеров биоинформатики, чтобы лучше понять полученные данные. Другой проблемой, связанной с этим протоколом, является создание крупномасштабных библиотек CRISPR. Подготовка этих обширных библиотек зависит от сравнительного увеличения ресурсов, необходимых для культивирования огромного числа клеток, необходимых для успешного скрининга многих нарушений.[8]
Параллельно с этими одноклеточными методами были разработаны другие подходы для реконструкции генетических путей с использованием секвенирования РНК всего организма. Эти методы используют единую совокупную статистику, называемую коэффициентом эпистаза в масштабе транскриптома, для определения пути реконструкции пути.[14] В отличие от статистической структуры методов, описанных выше, этот коэффициент может быть более устойчивым к шуму и интуитивно интерпретируется с точки зрения эпистаза Бейтсона. Этот подход был использован для определения нового состояния в жизненном цикле нематоды. C. elegans.[15]
Приложения
Perturb-seq или другие концептуально похожие протоколы могут использоваться для решения широкого круга биологических вопросов, и возможности применения этой технологии, вероятно, со временем будут расти. Три статьи по этой теме, опубликованные в выпуске журнала Journal Cell за декабрь 2016 г., продемонстрировали полезность этого метода, применив его к исследованию нескольких различных биологических функций. В статье «Perturb-Seq: рассечение молекулярных цепей с помощью масштабируемого одноклеточного РНК-профилирования объединенных генетических скринов» авторы использовали Perturb-seq для проведения нокаута факторы транскрипции связанный с иммунная реакция в сотнях тысяч клеток, чтобы исследовать клеточные последствия их инактивации.[2] Они также исследовали влияние факторов транскрипции на состояния клеток в контексте клеточный цикл. В исследовании под руководством UCSF, «Мультиплексная платформа CRISPR для скрининга одиночных клеток позволяет систематически анализировать развернутый белковый ответ», исследователи подавили несколько генов в каждой клетке, чтобы изучить развернутый белковый ответ (UPR) путь.[1] С помощью аналогичной методологии, но с использованием термина CRISP-seq вместо Perturb-seq, в статье «Рассечение иммунных цепей путем связывания CRISPR-Pooled Screens с Single-Cell RNA-Seq» был проведен эксперимент, подтверждающий концепцию, с использованием метода зондирования регуляторные пути, связанные с врожденный иммунитет у мышей.[4] Летальность каждого возмущения и эпистаз Анализы в ячейках с множественными возмущениями также исследовались в этих работах. Perturb-seq до сих пор использовался с очень небольшим количеством возмущений в эксперименте, но теоретически его можно масштабировать, чтобы охватить весь геном. Наконец, препринт октября 2016 г.[5] и последующий документ[3] продемонстрировать биоинформатическую реконструкцию сигнального пути Т-клеточного рецептора в Юркат ячейки на основе данных CROP-seq.
Хотя в этих публикациях эти протоколы использовались для ответа на сложные биологические вопросы, эту технологию также можно использовать в качестве валидационного анализа, чтобы гарантировать специфичность любого нокаута или нокаута на основе CRISPR; уровни экспрессии генов-мишеней, а также других генов могут быть измерены параллельно с разрешением отдельных клеток, чтобы определить, было ли возмущение успешным, и оценить эксперимент на предмет эффектов нецелевых. Кроме того, эти протоколы позволяют выполнять скрининг возмущений в гетерогенных тканях, получая при этом ответы на экспрессию генов, специфичных для клеточного типа.
Рекомендации
- ^ а б c d е ж грамм час Адамсон, Бритт; Норман, Томас М .; Йост, Марко; Чо, Мин Й .; Нуньес, Джеймс К .; Чен, Ювэнь; Виллалта, Жаклин Э .; Гилберт, Люк А .; Хорлбек, Макс А. (2016). «Мультиплексная платформа CRISPR для скрининга отдельных клеток позволяет систематически анализировать развернутый белковый ответ». Клетка. 167 (7): 1867–1882. E21. Дои:10.1016 / j.cell.2016.11.048. ЧВК 5315571. PMID 27984733.
- ^ а б c d е ж Диксит, Атрей; Парнас, Орен; Ли, Бию; Чен, Дженни; Фулько, Чарльз П .; Джерби-Арнон, Ливнат; Марьянович, Неманья Д .; Дионн, Даниэль; Беркс, Тайлер (2016). "Perturb-Seq: разделение молекулярных цепей с помощью масштабируемого профилирования одноклеточной РНК объединенных генетических скринингов". Клетка. 167 (7): 1853–1866.e17. Дои:10.1016 / j.cell.2016.11.038. ЧВК 5181115. PMID 27984732.
- ^ а б c d Датлингер, Пол; Рендейро, Андре Ф; Шмидл, Кристиан; Краусгрубер, Томас; Трэкслер, Питер; Клугаммер, Йоханна; Шустер, Линда С; Кучлер, Амели; Альпар, Донат (2017). «Объединенный CRISPR-скрининг со считыванием одноклеточного транскриптома». Природные методы. 14 (3): 297–301. Дои:10.1038 / nmeth.4177. ЧВК 5334791. PMID 28099430.
- ^ а б c d Джайтин, Диего Адхемар; Вайнер, Ассаф; Йофе, Идо; Лара-Астиасо, Давид; Керен-Шауль, Хадас; Давид, Эяль; Саламе, Томер Меир; Танай, Амос; Ауденаарден, Александр ван (2016). «Рассечение иммунных цепей путем связывания CRISPR-Pooled Screens с одноклеточной RNA-Seq». Клетка. 167 (7): 1883–1896.e15. Дои:10.1016 / j.cell.2016.11.039. PMID 27984734.
- ^ а б Датлингер, Пол; Шмидл, Кристиан; Рендейро, Андре Ф .; Трэкслер, Питер; Клугаммер, Йоханна; Шустер, Линда; Бок, Кристоф (27.10.2016). «Объединенный CRISPR-скрининг со считыванием одноклеточного транскриптома». bioRxiv 10.1101/083774.
- ^ «Объединенный CRISPR-скрининг со считыванием одноклеточного транскриптома». crop-seq.computational-epigenetics.org. Получено 2017-05-30.
- ^ Ларсон, Мэтью H; Гилберт, Люк А; Ван, Сяово; Лим, Венделл А; Вайсман, Джонатан С; Ци, Лей С. (2013). «Интерференция CRISPR (CRISPRi) для последовательного контроля экспрессии генов». Протоколы природы. 8 (11): 2180–2196. Дои:10.1038 / nprot.2013.132. ЧВК 3922765. PMID 24136345.
- ^ а б c d Шалем, Офир; Sanjana, Neville E .; Хартениан, Элла; Ши, Си; Скотт, Дэвид А .; Mikkelsen, Tarjei S .; Хекл, Дирк; Эберт, Бенджамин Л .; Рут, Дэвид Э. (03.01.2014). «Нокаут-скрининг CRISPR-Cas9 в масштабе генома в клетках человека». Наука. 343 (6166): 84–87. Дои:10.1126 / science.1247005. HDL:1721.1/111576. ISSN 0036-8075. ЧВК 4089965. PMID 24336571.
- ^ Ван, Тим; Вэй, Дженни Дж .; Сабатини, Дэвид М .; Лендер, Эрик С. (2014-01-03). «Генетический скрининг в клетках человека с использованием системы CRISPR-Cas9». Наука. 343 (6166): 80–84. Дои:10.1126 / science.1246981. ISSN 0036-8075. ЧВК 3972032. PMID 24336569.
- ^ Уилсон, Никола К .; Кент, Дэвид Дж .; Бюттнер, Флориан; Шехата, Мона; Macaulay, Iain C .; Калеро-Ньето, Фернандо Дж .; Кастильо, Мануэль Санчес; Oedekoven, Caroline A .; Диаманти, Евангелия (2015). «Комбинированный анализ функциональных характеристик отдельных клеток и экспрессии генов решает проблему неоднородности в популяциях стволовых клеток». Стволовая клетка. 16 (6): 712–724. Дои:10.1016 / j.stem.2015.04.004. ЧВК 4460190. PMID 26004780.
- ^ https://github.com/asncd/MIMOSCA
- ^ Бетчер, Майкл; Макманус, Майкл Т. (2015). «Выбор подходящего инструмента для работы: RNAi, TALEN или CRISPR». Молекулярная клетка. 58 (4): 575–585. Дои:10.1016 / j.molcel.2015.04.028. ЧВК 4441801. PMID 26000843.
- ^ Лю, Серена; Трапнелл, Коул (17 февраля 2016). «Секвенирование одноклеточного транскриптома: последние достижения и нерешенные проблемы». F1000 Исследования. 5: 182. Дои:10.12688 / f1000research.7223.1. ЧВК 4758375. PMID 26949524.
- ^ Анхелес-Альборес, Дэвид; Пакетт Робинсон, Карми; Уильямс, Брайан А; Уолд, Барбара Дж .; Штернберг, Пол В. (27 марта 2018 г.). «Реконструкция генетического пути многоклеточных животных с помощью транскриптомных измерений эпистаза». PNAS. 115 (13): E2930 – E2939. Дои:10.1073 / pnas.1712387115. ЧВК 5879656. PMID 29531064.
- ^ Анхелес-Альборес, Дэвид; Leighton, Daniel H.W .; Цоу, Тиффани; Khaw, Tiffany H .; Антошечкин Игорь; Штернберг, Пол В. (07.09.2017). "The Caenorhabditis elegans Состояние, подобное женскому: разделение транскриптомных эффектов старения и статуса спермы ». G3: Гены, геномы, генетика. 115 (9): 2969–2977. Дои:10.1534 / g3.117.300080. ЧВК 5592924. PMID 28751504.