Запрограммированная гибель клеток - Programmed cell death
Запрограммированная гибель клеток (PCD; иногда упоминается как клеточное самоубийство[1]) это смерть из ячейка в результате событий внутри ячейки, таких как апоптоз или аутофагия.[2][3] PCD проводится в биологический процесс, что обычно дает преимущество во время жизненный цикл. Например, дифференциация пальцев рук и ног в развивающемся человеческом эмбрионе происходит потому, что клетки между пальцами апоптоз; в результате цифры разделены. PCD выполняет основные функции во время обоих завод и животное развитие тканей.
Апоптоз и аутофагия обе формы запрограммированной гибели клеток.[4] Некроз - это смерть клетки, вызванная внешними факторами, такими как травма или инфекция, и происходит в нескольких различных формах. Некроз долгое время считался нефизиологическим процессом, возникающим в результате инфекции или травмы.[4] но в 2000-х годах форма запрограммированного некроза, называемая некроптоз,[5] была признана альтернативной формой запрограммированной гибели клеток. Предполагается, что некроптоз может служить резервом клеточной гибели для апоптоза, когда передача сигналов апоптоза блокируется эндогенными или экзогенными факторами, такими как вирусы или мутации. Совсем недавно были обнаружены и другие типы регулируемого некроза, которые разделяют несколько сигнальных событий с некроптозом и апоптозом.[6]
История
Концепция «запрограммированной смерти клеток» была использована Локшин И Уильямс[7] в 1964 г. по отношению к насекомое развитие тканей за восемь лет до изобретения «апоптоза». Однако термин PCD был источником путаницы, и Дюран и Рэмси[8] разработали концепцию, предоставив механистические и эволюционные определения. PCD стал общим термином, который относится ко всем различным типам гибели клеток, имеющим генетический компонент.
Первое понимание механизма пришло из изучения BCL2, продукт предполагаемого онкоген активирован хромосома транслокации часто встречается в фолликулярных лимфома. В отличие от других генов рака, которые способствуют рак Стимулируя пролиферацию клеток, BCL2 способствовал развитию рака, не давая клеткам лимфомы убивать себя.[9]
PCD был предметом все большего внимания и исследовательских усилий. Эта тенденция была подчеркнута наградой 2002 г. Нобелевская премия по физиологии и медицине к Сидней Бреннер (объединенное Королевство ), Х. Роберт Хорвиц (США) и Джон Э. Салстон (ВЕЛИКОБРИТАНИЯ).[10]
Типы
- Апоптоз или клеточная смерть типа I.
- Аутофагический или клеточная смерть типа II. (Цитоплазматический: характеризуется образованием больших вакуоли которые разъедают органеллы в определенной последовательности до разрушения ядро.)[11]
Апоптоз
Апоптоз это процесс запрограммированной гибели клеток (PCD), который может происходить в многоклеточные организмы.[12] Биохимический события приводят к характерным клеточным изменениям (морфология ) и смерть. Эти изменения включают пузыри, усадка ячейки, ядерный фрагментация конденсация хроматина, и хромосомный ДНК фрагментация. Сейчас считается, что - в контексте развития - клетки побуждаются к позитивному самоубийству, находясь в гомеостатическом контексте; отсутствие определенных факторов выживания может стать толчком к самоубийству. По-видимому, существуют некоторые различия в морфологии и, действительно, биохимии этих путей самоубийства; некоторые идут по пути «апоптоза», другие идут по более обобщенному пути к делеции, но оба обычно имеют генетическую и синтетическую мотивацию. Есть некоторые свидетельства того, что определенные симптомы «апоптоза», такие как активация эндонуклеаз, могут быть ложно индуцированы без участия генетического каскада, однако, по-видимому, истинный апоптоз и запрограммированная гибель клеток должны быть генетически опосредованы. Также становится ясно, что митоз и апоптоз каким-то образом переключаются или связаны, и что достигнутый баланс зависит от сигналов, полученных от соответствующих факторов роста или выживания.[13]
Аутофагия
Макроаутофагия, часто называемый аутофагия, это катаболический процесс, который приводит к аутофагосомный -лизосомный деградация массы цитоплазматический содержимое, аномальные белковые агрегаты, а также избыток или повреждение органеллы.
Аутофагия обычно активируется условиями питательное вещество депривация, но также была связана с физиологический а также патологический такие процессы, как развитие, дифференциация, нейродегенеративный болезни, стресс, инфекция и рак.
Механизм
Критическим регулятором индукции аутофагии является киназа mTOR, который при активации подавляет аутофагия а когда не активирован, продвигает его. Три связанных серин /треонин киназы, UNC-51-подобные киназы -1, -2 и -3 (ULK1, ULK2, UKL3), которые играют ту же роль, что и дрожжи Atg1, действовать после mTOR сложный. ULK1 и ULK2 образуют большой комплекс с млекопитающими гомолог продукта гена, связанного с аутофагией (Atg) (mAtg13), и каркасного белка FIP200. Комплекс PI3K класса III, содержащий hVps34, Беклин-1, p150 и Atg14-подобный белок или ген, связанный с устойчивостью к ультрафиолетовому облучению (UVRAG), необходим для индукции аутофагии.
В ПТГ гены контролировать аутофагосома формирование через ATG12 -ATG5 и LC3-II (ATG8 -II) комплексы. ATG12 сопряжен с ATG5 в убиквитин -подобная реакция, требующая ATG7 и ATG10. Конъюгат Atg12-Atg5 затем нековалентно взаимодействует с ATG16 с образованием большого комплекса. LC3 /ATG8 расщепляется на своем С-конце ATG4 протеаза для генерации цитозольного LC3-I. LC3-I конъюгирован с фосфатидилэтаноламином (PE) также в убиквитиноподобной реакции, для которой требуются Atg7 и Atg3. Липидированная форма LC3, известная как LC3-II, прикрепляется к мембране аутофагосомы.
Аутофагия и апоптоз связаны как положительно, так и отрицательно, и между ними существует значительная перекрестная помеха. В течение дефицит питательных веществ, аутофагия функционирует как механизм защиты выживания, однако аутофагия может привести к смерть клетки, процесс морфологически в отличие от апоптоз. Несколько проапоптотических сигналы, такие как TNF, ТРАССА, и FADD, также вызывают аутофагию. Дополнительно, Bcl-2 подавляет Беклин-1 -зависимая аутофагия, тем самым действуя как регулятор выживания и как антиаутофагический регулятор.
Другие типы
Помимо двух вышеупомянутых типов PCD, были обнаружены и другие пути.[14]Называется «неапоптотической запрограммированной гибелью клеток» (иликаспаза -независимая запрограммированная гибель клеток »или« некроптоз »), эти альтернативные пути к смерти столь же эффективны, как и апоптоз, и могут функционировать либо как резервные механизмы, либо как основной тип PCD.
Другие формы запрограммированной гибели клеток включают: Anoikis почти идентичен апоптозу, за исключением его индукции; ороговение форма гибели клеток исключительно для глаз; эксайтотоксичность; ферроптоз, железозависимая форма гибели клеток[15] и Валлеровское вырождение.
Некроптоз представляет собой запрограммированную форму некроза или воспалительной гибели клеток. Обычно некроз связан с незапрограммированной гибелью клеток в результате повреждения клеток или инфильтрации патогенами, в отличие от упорядоченной запрограммированной гибели клеток через апоптоз. Немозис это еще одна запрограммированная форма некроза, которая имеет место в фибробласты.[16]
Эриптоз это форма суицидального эритроцит смерть.[17]
Апонекроз представляет собой гибрид апоптоза и некроза и относится к неполному апоптотическому процессу, который завершается некрозом.[18]
Нетоз это процесс гибели клеток, вызванный СЕТИ.[19]
Параптоз это еще один тип неапоптотической гибели клеток, опосредованный MAPK через активацию IGF-1. Характеризуется внутриклеточным образованием вакуолей и набуханием митохондрий.[20]
Пироптоз, воспалительный тип гибели клеток, однозначно опосредуется каспаза 1, фермент, не участвующий в апоптозе в ответ на инфекцию определенными микроорганизмами.[20]
Клетки растений подвергаются определенным процессам PCD, подобным гибели аутофагических клеток. Однако некоторые общие черты PCD в высокой степени сохраняются как у растений, так и у многоклеточных.
Атрофические факторы
Атрофический фактор - это сила, вызывающая ячейка к умри. Только естественные силы, воздействующие на клетку, считаются атрофическими факторами, тогда как, например, агенты механического или химического воздействия или лизиса клетки не считаются атрофическими факторами.[кем? ] Распространенными типами атрофических факторов являются:[21]
- Сниженная нагрузка
- Потеря иннервации
- Снижение кровоснабжения
- Неадекватное питание
- Утрата эндокринный стимуляция
- Старость
- Сжатие
Роль в развитии нервной системы

Первоначальное расширение развивающейся нервная система уравновешивается удалением нейронов и их отростков.[22] В процессе развития нервной системы почти 50% развивающихся нейронов естественным образом удаляются в результате запрограммированной гибели клеток (PCD).[23] PCD в нервной системе был впервые обнаружен в 1896 году Джоном Бирдом.[24] С тех пор было предложено несколько теорий для понимания его биологического значения во время нейронное развитие.[25]
Роль в нервном развитии
PCD в развивающейся нервной системе наблюдается как в пролиферирующих, так и в постмитотических клетках.[22] Одна теория предполагает, что PCD - это адаптивный механизм, регулирующий количество клетки-предшественники. У людей PCD в клетках-предшественниках начинается на 7 неделе гестации и сохраняется до первого триместра.[26] Этот процесс гибели клеток был идентифицирован в зародышевых областях кора головного мозга, мозжечок, таламус, мозговой ствол, и спинной мозг среди других регионов.[25] На 19–23 неделях гестации в постмитотических клетках наблюдается PCD.[27] Преобладающей теорией, объясняющей это наблюдение, является нейротрофическая теория, согласно которой PCD требуется для оптимизации связи между нейронами и их афферентными входами и эфферентными мишенями.[25] Другая теория предполагает, что PCD развития в нервной системе происходит, чтобы исправить ошибки в нейронах, которые мигрировали эктопически, иннервируют неправильные цели или имеют аксоны что пошли наперекосяк при поиске пути.[28] Возможно, что PCD во время развития нервной системы выполняет различные функции, определяемые стадией развития, типом клеток и даже видами.[25]
Нейротрофическая теория
Нейротрофическая теория - это ведущая гипотеза, используемая для объяснения роли запрограммированной гибели клеток в развивающейся нервной системе.[29]. Он постулирует, что для обеспечения оптимальной иннервации мишеней сначала производится избыток нейронов, которые затем конкурируют за ограниченное количество защитных нейротрофические факторы и только часть выживает, в то время как другие умирают в результате запрограммированной гибели клеток.[26] Кроме того, теория утверждает, что предопределенные факторы регулируют количество выживших нейронов, а размер популяции иннервирующих нейронов напрямую коррелирует с влиянием их целевого поля.[30]
Основная идея о том, что клетки-мишени секретируют привлекательные или индуцирующие факторы и что их шишки есть хемотаксический чувствительность была впервые высказана Сантьяго Рамон-и-Кахаль в 1892 г.[31] Кахал представил эту идею как объяснение «разумной силы», которую аксоны принимают при нахождении своей цели, но признал, что у него нет эмпирических данных.[31] Теория стала более привлекательной, когда экспериментальные манипуляции с мишенями аксонов привели к гибели всех иннервирующих нейронов. Это привело к развитию концепции регуляции, производной от мишени, которая стала основным принципом нейротрофической теории.[32][33] Эксперименты, которые еще больше подтвердили эту теорию, привели к идентификации первого нейротрофического фактора, фактор роста нервов (NGF).[34]
Периферическая и центральная нервная система
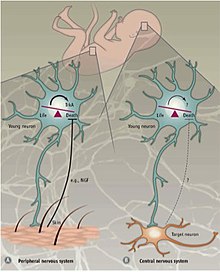
Различные механизмы регулируют PCD в периферическая нервная система (PNS) по сравнению с Центральная нервная система (ЦНС). В ПНС иннервация мишени пропорциональна количеству высвобождаемых мишенью нейротрофических факторов NGF и NT3.[35][36] Экспрессия рецепторов нейротрофина, TrkA и TrkC, достаточно, чтобы вызвать апоптоз в отсутствие их лиганды.[23] Таким образом, предполагается, что PCD в PNS зависит от высвобождения нейротрофических факторов и, таким образом, следует концепции нейротрофической теории.
Запрограммированная гибель клеток в ЦНС не зависит от внешних факторов. факторы роста но вместо этого полагается на внутренние сигналы. в неокортекс, соотношение возбуждающего и тормозящего 4: 1 интернейроны поддерживается аппаратом апоптоза, который кажется независимым от окружающей среды.[36] Подтверждающие доказательства были получены в результате эксперимента, в котором предшественники интернейронов были либо трансплантированы в неокортекс мыши, либо культивированы. in vitro.[37] Пересаженные клетки погибли в возрасте двух недель, в том же возрасте, в котором эндогенные интернейроны подвергаются апоптозу. Независимо от размера трансплантата, доля клеток, подвергшихся апоптозу, оставалась постоянной. Кроме того, нарушение TrkB рецептор для нейротрофический фактор головного мозга (Bdnf) не влияли на гибель клеток. Также было показано, что у мышей нулевой проапоптотический фактор Bax (Bcl-2-ассоциированный X-белок) выживал больший процент интернейронов по сравнению с мышами дикого типа.[37] Вместе эти находки указывают на то, что запрограммированная гибель клеток в ЦНС частично использует передачу сигналов, опосредованную Bax, и не зависит от BDNF и окружающей среды. Механизмы апоптоза в ЦНС все еще недостаточно изучены, но считается, что апоптоз интернейронов является самоавтономным процессом.
Развитие нервной системы при ее отсутствии
Запрограммированная гибель клеток может быть уменьшена или устранена в развивающейся нервной системе путем целенаправленной делеции проапоптотических генов или сверхэкспрессии антиапоптотических генов. Отсутствие или уменьшение PCD может вызвать серьезные анатомические аномалии, но также может привести к минимальным последствиям в зависимости от целевого гена, популяции нейронов и стадии развития.[25] Избыточная пролиферация клеток-предшественников, которая приводит к грубым аномалиям головного мозга, часто приводит к летальному исходу, как показано на каспаза-3 или каспаза-9 нокаутные мыши которые развиваются экзэнцефалия в передний мозг.[38][39] Ствол головного мозга, спинной мозг и периферические ганглии этих мышей развиваются нормально, однако, это предполагает участие каспасы при PCD во время развития зависит от области мозга и типа клеток.[40] Нокаут или ингибирование фактора активации апоптотической протеазы 1 (APAF1 ), также приводит к порокам развития и повышенной эмбриональной летальности.[41][42][43] Манипуляции с белками-регуляторами апоптоза Bcl-2 и Bax (сверхэкспрессия Bcl-2 или делеция Bax) вызывает увеличение числа нейронов в определенных областях нервной системы, таких как сетчатка, ядро тройничного нерва, мозжечок и спинной мозг.[44][45][46][47][48][49][50] Однако PCD нейронов из-за делеции Bax или сверхэкспрессии Bcl-2 не приводит к заметным морфологическим или поведенческим аномалиям у мышей. Например, мыши со сверхэкспрессией Bcl-2 в целом имеют нормальные моторные навыки и зрение и демонстрируют нарушения только в сложных формах поведения, таких как обучение и тревожность.[51][52][53] Нормальное поведенческое фенотипы этих мышей предполагают, что может быть задействован адаптивный механизм для компенсации избытка нейронов.[25]
Беспозвоночные и позвоночные

Изучение PCD у различных видов важно для понимания эволюционной основы и причины апоптоза в развитии нервной системы. Во время разработки беспозвоночный нервная система, PCD играет разные роли у разных видов[54]. Сходство механизма асимметричной гибели клеток в нематода и пиявка указывает на то, что PCD может иметь эволюционное значение в развитии нервной системы.[55] У нематод PCD происходит в первый час развития, что приводит к элиминации 12% негонадных клеток, включая нейрональные клоны.[56] Смерть клетки в членистоногие возникает сначала в нервной системе, когда эктодерма клетки дифференцируются, и одна дочерняя клетка становится нейробласт а другой подвергается апоптозу.[57] Кроме того, гибель клеток, нацеленных на пол, приводит к разной иннервации нейронов определенных органов у мужчин и женщин.[58] В Дрозофила, PCD играет важную роль в сегментации и спецификации во время разработки.
В отличие от беспозвоночных, механизм запрограммированной гибели клеток более консервативен у позвоночные. Обширные исследования, проведенные на различных позвоночных, показывают, что PCD нейронов и глия происходит в большинстве частей нервной системы во время развития. Это наблюдалось до и во время синаптогенез в центральной нервной системе, а также в периферической нервной системе.[25] Однако между видами позвоночных есть несколько различий. Например, млекопитающие демонстрируют обширное ветвление с последующим PCD в сетчатке, в то время как птицы этого не делают.[59] Хотя синаптическое совершенствование в системах позвоночных в значительной степени зависит от PCD, другие эволюционные механизмы также играют роль.[25]
В растительной ткани
Запрограммированная гибель клеток растений имеет ряд молекулярных сходств с животными. апоптоз, но есть и отличия, наиболее очевидным из которых является наличие клеточная стенка и отсутствие иммунная система который удаляет осколки мертвой клетки. Вместо иммунного ответа умирающая клетка синтезирует вещества, чтобы разрушиться, и помещает их в вакуоль который разрывается, когда клетка умирает.[60]
В «APL» регулирует идентичность сосудистой ткани в Арабидопсис ",[61] Мартин Бонке и его коллеги заявили, что одна из двух транспортных систем дальнего следования в сосудистые растения, ксилема, состоит из нескольких типов клеток, дифференциация которых включает отложение сложных клеточная стенка утолщения и запрограммированной гибели клеток ». Авторы подчеркивают, что продукты PCD растений играют важную структурную роль.
Основные морфологические и биохимические признаки ПХД сохранены как у растений, так и у животных. королевства.[62] Определенные типы растительных клеток реализуют уникальные программы гибели клеток. Они имеют общие черты с апоптозом животных - например, ядерная ДНК деградация - но у них есть и свои особенности, такие как ядерный деградация, вызванная крахом вакуоль в трахея элементы ксилемы.[63]
Яннеке Балк и Кристофер Дж. Ливер из Департамента Науки о растениях, Оксфордский университет, провели исследование мутаций в митохондриальный геном из подсолнух клетки. Результаты этого исследования показывают, что митохондрии играют такую же ключевую роль в PCD сосудистых растений, как и в других эукариотический клетки.[64]
PCD в пыльце предотвращает инбридинг
В течение опыление, растения обеспечивают соблюдение несовместимость с самим собой (SI) как важное средство предотвращения самооплодотворение. Исследования по кукурузный мак (Papaver Rhoeas) показал, что белки в пестик на котором пыльца земли, взаимодействуют с пыльцой и вызывают ПКС в несовместимых (т.е. я) пыльца. Исследователи Стивен Г. Томас и Вероника Э. Франклин-Тонг также обнаружили, что реакция включает быстрое подавление пыльцевая трубка рост, затем PCD.[65]
В формах слизи
Социальная слизь Dictyostelium discoideum имеет особенность принятия хищного амеба -подобное поведение в своем одноклеточный форма или объединение в мобильный слизняк -подобная форма при распылении споры что даст рождение следующего поколение.[66]
Стебель состоит из мертвых клеток, которые подверглись типу PCD, который имеет много общих черт аутофагической гибели клеток: массивные вакуоли, образующиеся внутри клеток, в некоторой степени хроматин конденсат, но нет Фрагментация ДНК.[67] Структурная роль остатков, оставленных мертвыми клетками, напоминает продукты PCD в растительной ткани.
D. discoideum слизь, часть ветки, которая могла появиться из эукариотический предки о миллиард лет до настоящего. Кажется, что они возникли после предков зеленые растения и предки грибы и животные дифференцировались. Но, помимо своего места в эволюционной дерево, факт, что PCD наблюдалась в скромных, простых, шести-хромосома D. discoideum имеет дополнительное значение: он позволяет изучить путь развития PCD, который не зависит от каспаз, характерных для апоптоза.[68]
Эволюционное происхождение митохондриального апоптоза
Возникновение запрограммированной гибели клеток в протисты возможно,[69][70] но это остается спорным. Некоторые относят смерть этих организмов к нерегулируемой гибели клеток, подобной апоптозу.[71][72]
Биологи давно подозревали, что митохондрии возник из бактерии которые были включены как эндосимбионты («живущие вместе внутри») более крупных эукариотических клеток. Это было Линн Маргулис кто с 1967 года отстаивал это теория, который с тех пор получил широкое распространение.[73] Самый убедительный доказательства для этой теории является тот факт, что митохондрии обладают собственными ДНК и оснащены гены и репликация аппарат.
Эта эволюционный шаг был бы рискованным для примитивных эукариотических клеток, которые начали поглощать производство энергии бактерии, а также опасный шаг для предков митохондрий, которые начали вторгаться в их протоэукариотические хозяева. Этот процесс очевиден и сегодня, между человек белый клетки крови и бактерии. В большинстве случаев вторгшиеся бактерии уничтожаются лейкоцитами; однако это не редкость для химическая война ведется прокариоты чтобы добиться успеха, с последствиями, известными как инфекция в результате ущерба.
Одно из этих редких эволюционных событий, два миллиарда лет до настоящего времени позволял некоторым эукариотам и прокариотам, производящим энергию, сосуществовать и получать взаимную выгоду от своего симбиоз.[74]
Митохондриальные эукариотические клетки живут, балансируя между жизнь и смерть, потому что митохондрии все еще сохраняют свой репертуар молекулы что может спровоцировать самоубийство клетки.[75] Неясно, почему аппарат апоптоза сохраняется у современных одноклеточных организмов. Теперь этот процесс должен происходить только тогда, когда он запрограммирован.[76] в клетки (например, обратная связь от соседей, стресс или Повреждение ДНК ), высвобождение митохондрий каспаза активаторы, вызывающие гибель клеток биохимический каскад. Таким образом, клеточное самоубийство механизм теперь имеет решающее значение для всей нашей жизни.
Запрограммированная смерть целых организмов
Клиническое значение
ABL
Было обнаружено, что онкоген BCR-ABL участвует в развитии рак в людях.[77]
c-Myc
c-Myc участвует в регуляции апоптоза благодаря своей роли в подавлении Bcl-2 ген. Его роль в нарушенном росте тканей.[77]
Метастаз
А молекулярный Характерной чертой метастатических клеток является измененная экспрессия нескольких апоптотических генов.[77]
Смотрите также
- Аноикис
- Фактор, вызывающий апоптоз
- Апоптоз против Псевдоапоптоз
- Апоптосома
- Апоптотическая фрагментация ДНК
- Автолиз (биология)
- Аутофагия
- Автощизис
- Bcl-2
- Агонист смерти взаимодействующего домена BH3 (BID)
- Calpains
- Каспасы
- Повреждение клеток
- Корнификация
- Цитохром с
- Цитотоксичность
- Гомолог Diablo
- Entosis
- Эксайтотоксичность
- Ферроптоз
- Инфламмасома
- Митохондриальная проницаемость переходной поры
- Митотическая катастрофа
- Некробиология
- Некроптоз
- Некроз
- модулятор апоптоза с повышенной регуляцией p53 (PUMA)
- Параптоз
- Parthanatos
- Пироптоз
- Киназы RIP
- Валлеровское вырождение
Примечания и ссылки
- Шривастава Р.Э. в молекулярных механизмах (Humana Press, 2007).
- Кирзенбаум, А. Л. и Трес, Л. Л. (под редакцией Мадлен Хайд) (ELSEVIER SAUNDERS, Филадельфия, 2012).
- ^ Рафф, М. (12 ноября 1998 г.). «Самоубийство клетки для новичков». Природа. 396 (6707): 119–22. Дои:10.1038/24055. ISSN 0028-0836. PMID 9823889. S2CID 4341684.
- ^ Энгельберг-Кулька Х, Амитаи С., Колодкин-Гал И., Хазан Р. (2006). «Бактериальная программируемая клеточная смерть и многоклеточное поведение бактерий». PLOS Genetics. 2 (10): e135. Дои:10.1371 / journal.pgen.0020135. ЧВК 1626106. PMID 17069462.
- ^ Грин, Дуглас (2011). Средство для достижения цели. Нью-Йорк: Лаборатория издательства Колд-Спринг-Харбор. ISBN 978-0-87969-887-4.
- ^ а б Кирзенбаум, Авраам (2012). Гистология и клеточная биология - Введение в патологию. Филадельфия: ELSEVIER SAUNDERS.
- ^ Дегтерев Алексей; Хуанг, Чжихун; Бойс, Майкл; Ли, Яцяо; Джагтап, Пракаш; Мидзусима, Нобору; Куни, Грегори Д.; Митчисон, Тимоти Дж .; Московиц, Майкл А. (01.07.2005). «Химический ингибитор неапоптотической гибели клеток с терапевтическим потенциалом при ишемическом повреждении головного мозга». Природа Химическая Биология. 1 (2): 112–119. Дои:10.1038 / nchembio711. ISSN 1552-4450. PMID 16408008. S2CID 866321.
- ^ Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P (2014). «Регулируемый некроз: расширяющаяся сеть неапоптотических путей гибели клеток». Нат Рев Мол Cell Biol. 15 (2): 135–147. Дои:10.1038 / nrm3737. PMID 24452471. S2CID 13919892.
- ^ Локшин Р.А., Вильямс С.М. (1964). «Запрограммированная гибель клеток - II. Эндокринное усиление разрушения межсегментарных мышц тутового шелкопряда». Журнал физиологии насекомых. 10 (4): 643–649. Дои:10.1016/0022-1910(64)90034-4.
- ^ Дюран и Рэмси, Пьер М. и Грант (2019). «Природа запрограммированной гибели клеток» (PDF). Биологическая теория. 14: 30–41. Дои:10.1007 / s13752-018-0311-0. S2CID 91622808.
- ^ Vaux DL, Cory S, Adams JM (сентябрь 1988 г.). «Ген Bcl-2 способствует выживанию гемопоэтических клеток и взаимодействует с c-myc для иммортализации пре-B-клеток». Природа. 335 (6189): 440–2. Bibcode:1988Натура.335..440В. Дои:10.1038 / 335440a0. PMID 3262202. S2CID 23593952.
- ^ «Нобелевская премия по физиологии и медицине 2002 г.». Нобелевский фонд. 2002. Получено 2009-06-21.
- ^ Шварц Л.М., Смит С.В., Джонс М.Э., Осборн Б.А. (1993). «Все ли запрограммированные смерти клеток происходят через апоптоз?». PNAS. 90 (3): 980–4. Bibcode:1993ПНАС ... 90..980С. Дои:10.1073 / пнас.90.3.980. ЧВК 45794. PMID 8430112.; а более свежий взгляд см. Бурш В., Эллингер А., Гернер С., Фрёвайн Ю., Шульте-Герман Р. (2000). «Запрограммированная гибель клеток (PCD). Апоптоз, аутофагический PCD или другие?». Летопись Нью-Йоркской академии наук. 926 (1): 1–12. Bibcode:2000НЯСА.926 .... 1Б. Дои:10.1111 / j.1749-6632.2000.tb05594.x. PMID 11193023.
- ^ Грин, Дуглас (2011). Средство для достижения цели. Нью-Йорк: Лаборатория Прессы Колд-Спринг-Харбор. ISBN 978-0-87969-888-1.
- ^ Д. Боуэн, Айвор (1993). "Интернэшнл клеточной биологии 17". Cell Biology International. 17 (4): 365–380. Дои:10.1006 / cbir.1993.1075. PMID 8318948. S2CID 31016389. Архивировано из оригинал на 2014-03-12. Получено 2012-10-03.
- ^ Кремер Г, Мартин SJ (2005). «Каспазонезависимая гибель клеток». Природа Медицина. 11 (7): 725–30. Дои:10,1038 / нм 1263. PMID 16015365. S2CID 8264709.
- ^ Диксон Скотт Дж .; Лемберг Кэтрин М .; Lamprecht Michael R .; Скута Рашид; Зайцева Елена М .; Глисон Кэролайн Э .; Patel Darpan N .; Bauer Andras J .; Cantley Alexandra M .; и другие. (2012). «Ферроптоз: железозависимая форма неапоптотической гибели клеток». Ячейка. 149 (5): 1060–1072. Дои:10.1016 / j.cell.2012.03.042. ЧВК 3367386. PMID 22632970.
- ^ Юзеф Бизик; Эско Канкури; Ари Ристимяки; Ален Тайеб; Хейкки Вапаатало; Вернер Любиц; Антти Вахери (2004). «Контакты клетки вызывают запрограммированный некроз и индуцируют экспрессию циклооксигеназы-2». Гибель клеток и дифференциация. 11 (2): 183–195. Дои:10.1038 / sj.cdd.4401317. PMID 14555963.
- ^ Lang, F; Lang, KS; Ланг, Пенсильвания; Huber, SM; Видер, Т. (2006). «Механизмы и значение эриптоза». Антиоксиданты и редокс-сигналы. 8 (7–8): 1183–92. Дои:10.1089 / ars.2006.8.1183. PMID 16910766.
- ^ Формигли, L; и другие. (2000). «апонекроз: морфологические и биохимические исследования синкретического процесса гибели клеток, разделяющего апоптоз и некроз». Журнал клеточной физиологии. 182 (1): 41–49. Дои:10.1002 / (sici) 1097-4652 (200001) 182: 1 <41 :: aid-jcp5> 3.0.co; 2-7. PMID 10567915.
- ^ Фадини, врач общей практики; Menegazzo, L; Скаттолини, V; Gintoli, M; Альбиеро, М; Авогаро, А (25 ноября 2015 г.). «Взгляд на НЕТоз при диабете и кардиометаболических расстройствах». Питание, обмен веществ и сердечно-сосудистые заболевания: NMCD. 26 (1): 1–8. Дои:10.1016 / j.numecd.2015.11.008. PMID 26719220.
- ^ а б Росс, Майкл (2016). Гистология: текст и атлас (7-е изд.). п. 94. ISBN 978-1451187427.
- ^ Глава 10: Все игроки на одной сцене В архиве 2013-05-28 в Wayback Machine из PsychEducation.org
- ^ а б Тау, ГЗ (2009). «Нормальное развитие мозговых цепей». Нейропсихофармакология. 35 (1): 147–168. Дои:10.1038 / npp.2009.115. ЧВК 3055433. PMID 19794405.
- ^ а б Деккерс, депутат (2013). «Смерть развивающихся нейронов: новые идеи и значение для подключения». Журнал клеточной биологии. 203 (3): 385–393. Дои:10.1083 / jcb.201306136. ЧВК 3824005. PMID 24217616.
- ^ Оппенгейм, Р.В. (1981). Гибель нейрональных клеток и некоторые связанные с ней регрессивные явления во время нейрогенеза: выборочный исторический обзор и отчет о прогрессе. В исследованиях в области нейробиологии развития: очерки в честь Виктора Гамбургера: Oxford University Press. С. 74–133.
- ^ а б c d е ж г час Бусс, Р.Р. (2006). «Адаптивные роли запрограммированной гибели клеток в процессе развития нервной системы». Ежегодный обзор нейробиологии. 29: 1–35. Дои:10.1146 / annurev.neuro.29.051605.112800. PMID 16776578.
- ^ а б Де ла Роса, EJ; Де Пабло, Ф (23 октября 2000 г.). «Смерть клеток в раннем нервном развитии: за пределами нейротрофической теории». Тенденции в неврологии. 23 (10): 454–458. Дои:10.1016 / s0166-2236 (00) 01628-3. PMID 11006461. S2CID 10493404.
- ^ Лосси, L; Мериги, А. (апрель 2003 г.). «In vivo клеточные и молекулярные механизмы апоптоза нейронов в ЦНС млекопитающих». Прогресс в нейробиологии. 69 (5): 287–312. Дои:10.1016 / s0301-0082 (03) 00051-0. PMID 12787572. S2CID 27052883.
- ^ Финли, Б.Л. (1989). «Контроль количества клеток в развивающейся зрительной системе млекопитающих». Прогресс в нейробиологии. 32 (3): 207–234. Дои:10.1016/0301-0082(89)90017-8. PMID 2652194. S2CID 2788103.
- ^ Ямагути, Ёсифуми; Миура, Масаюки (23 февраля 2015). «Запрограммированная смерть клетки в нейроразвитии». Клетка развития. 32 (4): 478–490. Дои:10.1016 / j.devcel.2015.01.019. ISSN 1534-5807. PMID 25710534.
- ^ Рубинштейн, Джон; Пасько Ракич (2013). "Регуляция выживания нейронов нейротрофинами в развивающейся периферической нервной системе". Определение паттерна и типов клеток в развивающейся ЦНС и ПНС: комплексная нейробиология развития. Академическая пресса. ISBN 978-0-12-397348-1.
- ^ а б Константино, Сотело (2002). Хемотаксическая гипотеза Кахала: столетие позади. Прогресс в исследованиях мозга. 136. С. 11–20. Дои:10.1016 / s0079-6123 (02) 36004-7. ISBN 9780444508157. PMID 12143376.
- ^ Оппенгейм, Рональд (1989). «Нейротрофическая теория и естественная смерть мотонейронов». Тенденции в неврологии. 12 (7): 252–255. Дои:10.1016/0166-2236(89)90021-0. PMID 2475935. S2CID 3957751.
- ^ Деккерс, депутат; Николетопулу, В; Барде Ю.А. (11 ноября 2013 г.). «Клеточная биология в нейробиологии: смерть развивающихся нейронов: новые идеи и последствия для связи». J Cell Biol. 203 (3): 385–393. Дои:10.1083 / jcb.201306136. ЧВК 3824005. PMID 24217616.
- ^ Коуэн, WN (2001). «Виктор Гамбургер и Рита Леви-Монтальчини: путь к открытию фактора роста нервов». Ежегодный обзор нейробиологии. 24: 551–600. Дои:10.1146 / annurev.neuro.24.1.551. PMID 11283321. S2CID 6747529.
- ^ Велтман, Дж. К. (8 февраля 1987 г.). «Нобелевская премия по физиологии и медицине 1986 года присуждена за открытие факторов роста: Рита Леви-Монтальчини, доктор медицины, и Стэнли Коэн, доктор философии». Региональные слушания по аллергии Новой Англии. 8 (1): 47–8. Дои:10.2500/108854187779045385. PMID 3302667.
- ^ а б Деккерс, М. (5 апреля 2013 г.). «Запрограммированная смерть клетки в развитии нейронов». Наука. 340 (6128): 39–41. Bibcode:2013Наука ... 340 ... 39D. Дои:10.1126 / science.1236152. PMID 23559240. S2CID 206548254.
- ^ а б Саутвелл, Д. (Ноябрь 2012 г.). «Внутренне детерминированная гибель клеток развивающихся корковых интернейронов». Природа. 491 (7422): 109–115. Bibcode:2012Натура 491..109С. Дои:10.1038 / природа11523. ЧВК 3726009. PMID 23041929.
- ^ Куйда, К. (1998). «Снижение апоптоза и опосредованной цитохромом с активации каспазы у мышей, лишенных каспазы 9». Ячейка. 94 (3): 325–337. Дои:10.1016 / s0092-8674 (00) 81476-2. PMID 9708735. S2CID 8417446.
- ^ Куйда, К. (1996). «Снижение апоптоза в головном мозге и преждевременной летальности у мышей с дефицитом CPP32». Природа. 384 (6607): 368–372. Bibcode:1996Натура.384..368K. Дои:10.1038 / 384368a0. PMID 8934524. S2CID 4353931.
- ^ Оппенгейм, RW (2001). «Запрограммированная гибель клеток развивающихся нейронов млекопитающих после генетической делеции каспаз». Журнал неврологии. 21 (13): 4752–4760. Дои:10.1523 / JNEUROSCI.21-13-04752.2001. PMID 11425902.
- ^ Чеккони, Ф (1998). «Apaf1 (гомолог CED-4) регулирует запрограммированную гибель клеток в развитии млекопитающих». Ячейка. 94 (6): 727–737. Дои:10.1016 / s0092-8674 (00) 81732-8. PMID 9753320.
- ^ Хао, Z (2005). «Специфическое устранение апоптотических функций цитохрома с выявляет дифференциальную потребность в цитохроме с и Apaf-1 при апоптозе». Ячейка. 121 (4): 579–591. Дои:10.1016 / j.cell.2005.03.016. PMID 15907471. S2CID 4921039.
- ^ Йошида, H (1998). «Apaf1 необходим для митохондриальных путей апоптоза и развития мозга». Ячейка. 94 (6): 739–750. Дои:10.1016 / s0092-8674 (00) 81733-х. PMID 9753321. S2CID 1096066.
- ^ Бонфанти, Л. (1996). «Защита ганглиозных клеток сетчатки от естественной и вызванной аксотомией гибели клеток у неонатальных трансгенных мышей, сверхэкспрессирующих bcl-2». Журнал неврологии. 16 (13): 4186–4194. Дои:10.1523 / JNEUROSCI.16-13-04186.1996. PMID 8753880.
- ^ Мартину, JC (1994). «Сверхэкспрессия BCL-2 у трансгенных мышей защищает нейроны от естественной гибели клеток и экспериментальной ишемии». Нейрон. 13 (4): 1017–1030. Дои:10.1016/0896-6273(94)90266-6. PMID 7946326. S2CID 25546670.
- ^ Занджани, HS (1996). «Повышенное количество клеток Пуркинье мозжечка у мышей, сверхэкспрессирующих человеческий трансген bcl-2». Журнал вычислительной неврологии. 374 (3): 332–341. Дои:10.1002 / (sici) 1096-9861 (19961021) 374: 3 <332 :: aid-cne2> 3.0.co; 2-2. PMID 8906502.
- ^ Зуп, SL (2003). «Избыточная экспрессия bcl-2 снижает половые различия в количестве нейронов в головном и спинном мозге». Журнал неврологии. 23 (6): 2357–2362. Дои:10.1523 / JNEUROSCI.23-06-02357.2003. PMID 12657695.
- ^ Вентилятор, H (2001). «Устранение экспрессии Bax у мышей увеличивает количество клеток Пуркинье мозжечка, но не количество гранулярных клеток». Журнал вычислительной неврологии. 436 (1): 82–91. Дои:10.1002 / cne.1055.abs. PMID 11413548.
- ^ Мозингер, Огилви (1998). «Подавление онтогенетической гибели клеток сетчатки, но не дегенерации фоторецепторов у Bax-дефицитных мышей». Исследовательская офтальмология и визуализация. 39: 1713–1720.
- ^ Белый, FA (1998). «Широкое устранение естественной гибели нейронов у Bax-дефицитных мышей». Журнал неврологии. 18 (4): 1428–1439. Дои:10.1523 / JNEUROSCI.18-04-01428.1998. ЧВК 6792725. PMID 9454852.
- ^ Джанфранчески, Л. (1999). «Острота зрения поведения трансгенных мышей дикого типа и bcl2». Исследование зрения. 39 (3): 569–574. Дои:10.1016 / с0042-6989 (98) 00169-2. PMID 10341985. S2CID 5544203.
- ^ Ронди-Рейг, Л. (2002). «Умереть или не умирать, меняет ли это функцию? Поведение трансгенных мышей выявляет роль в гибели клеток в процессе развития». Бюллетень исследований мозга. 57 (1): 85–91. Дои:10.1016 / s0361-9230 (01) 00639-6. PMID 11827740. S2CID 35145189.
- ^ Ронди-Рейг, Л. (2001). «Трансгенные мыши с нейрональной сверхэкспрессией гена bcl-2 демонстрируют нарушения навигации при выполнении водных задач». Неврология. 104 (1): 207–215. Дои:10.1016 / s0306-4522 (01) 00050-1. PMID 11311543. S2CID 30817916.
- ^ Басс, Роберт Р .; Солнце, Вун; Оппенгейм, Рональд В. (21 июля 2006 г.). «Адаптивные роли запрограммированной гибели клеток в процессе развития нервной системы». Ежегодный обзор нейробиологии. 29 (1): 1–35. Дои:10.1146 / annurev.neuro.29.051605.112800. ISSN 0147-006X. PMID 16776578.
- ^ Салстон, Дж. Э. (1980). «Самец Caenorhabditis elegans: постэмбриональное развитие негонадальных структур». Биология развития. 78 (2): 542–576. Дои:10.1016/0012-1606(80)90352-8. PMID 7409314.
- ^ Sulston2, JE (1983). «Эмбриональная клеточная линия нематоды Caenorhabditis elegans». Биология развития. 100 (1): 64–119. Дои:10.1016/0012-1606(83)90201-4. PMID 6684600.
- ^ Доу, Cq (1985). «Развитие и сегментарные различия в структуре клеток-предшественников нейронов». Журнал биологии развития. 111 (1): 193–205. Дои:10.1016/0012-1606(85)90445-2. PMID 4029506.
- ^ Гибултович, JM (1984). «Половая дифференциация в терминальном ганглии моли Manduca sexta: роль половой гибели нейронов». Журнал сравнительной неврологии. 226 (1): 87–95. Дои:10.1002 / cne.902260107. PMID 6736297. S2CID 41793799.
- ^ Кук, Б. (1998). «Гибель нейронов в процессе развития не является универсальным явлением среди типов клеток сетчатки куриного эмбриона». Журнал сравнительной неврологии. 396 (1): 12–19. Дои:10.1002 / (sici) 1096-9861 (19980622) 396: 1 <12 :: aid-cne2> 3.0.co; 2-l. PMID 9623884.
- ^ Collazo C, Chacón O, Borrás O (2006). «Запрограммированная гибель клеток растений напоминает апоптоз животных» (PDF). Biotecnología Aplicada. 23: 1–10. Архивировано из оригинал (PDF) 14 марта 2012 г.
- ^ Бонке М., Титамади С., Мяхёнен А.П., Хаузер М.Т., Хелариутта Ю. (2003). «APL регулирует идентичность сосудистой ткани у Arabidopsis». Природа. 426 (6963): 181–6. Bibcode:2003Натура 426..181Б. Дои:10.1038 / природа02100. PMID 14614507. S2CID 12672242.
- ^ Соломон М., Беленги Б., Делледон М., Менахем Э., Левин А. (1999). «Участие цистеиновых протеаз и генов ингибиторов протеаз в регуляции запрограммированной гибели клеток у растений». Растительная клетка. 11 (3): 431–44. Дои:10.2307/3870871. JSTOR 3870871. ЧВК 144188. PMID 10072402. См. Также статьи по теме в Растительная клетка онлайн
- ^ Ито Дж, Фукуда Х (2002). «ZEN1 - ключевой фермент деградации ядерной ДНК во время запрограммированной гибели трахеарных элементов». Растительная клетка. 14 (12): 3201–11. Дои:10.1105 / tpc.006411. ЧВК 151212. PMID 12468737.
- ^ Балк Дж., Ливер CJ (2001). «Митохондриальная мутация PET1-CMS в подсолнечнике связана с преждевременной запрограммированной смертью клеток и высвобождением цитохрома c». Растительная клетка. 13 (8): 1803–18. Дои:10.1105 / tpc.13.8.1803. ЧВК 139137. PMID 11487694.
- ^ Томас С.Г., Франклин-Тонг В.Э. (2004). «Самонесовместимость вызывает запрограммированную гибель клеток в пыльце папавера». Природа. 429 (6989): 305–9. Bibcode:2004Натура 429..305Т. Дои:10.1038 / природа02540. PMID 15152254. S2CID 4376774.
- ^ Креспи Б., Спрингер С. (2003). «Экология. Социальные плесневые грибки встречаются с ними». Наука. 299 (5603): 56–7. Дои:10.1126 / science.1080776. PMID 12511635. S2CID 83917994.
- ^ Левро Дж. П., Адам М., Лучани М. Ф., де Шастелье С., Блэнтон Р. Л., Гольштейн П. (2003). «Гибель клеток Dictyostelium: раннее появление и исчезновение сильно поляризованных лопаточных клеток». Журнал клеточной биологии. 160 (7): 1105–14. Дои:10.1083 / jcb.200212104. ЧВК 2172757. PMID 12654899.
- ^ Ройзен-Буффе К., Лучани М.Ф., Кляйн Г., Левро Дж. П., Адам М., Гольштейн П. (2004). «Смерть развивающейся клетки в dictyostelium не требует паракаспазы». Журнал биологической химии. 279 (12): 11489–94. Дои:10.1074 / jbc.M312741200. PMID 14681218.
- ^ Депонте, М. (2008). «Запрограммированная гибель клеток у протистов». Biochimica et Biophysica Acta (BBA) - Исследование молекулярных клеток. 1783 (7): 1396–1405. Дои:10.1016 / j.bbamcr.2008.01.018. PMID 18291111.
- ^ Kaczanowski S, Sajid M and Reece S E 2011 Эволюция апоптозоподобной запрограммированной гибели клеток у одноклеточных простейших паразитов Parasites Vectors 4 44
- ^ Proto, W. R .; Coombs, G.H .; Моттрам, Дж. К. (2012). «Смерть клеток у паразитических простейших: регулируемая или случайная?» (PDF). Обзоры природы Микробиология. 11 (1): 58–66. Дои:10.1038 / nrmicro2929. PMID 23202528. S2CID 1633550. Архивировано из оригинал (PDF) на 2016-03-03. Получено 2014-11-14.
- ^ Шимон Качановски; Мохаммед Саджид; Сара Э. Рис (2011). «Эволюция апоптозоподобной запрограммированной гибели клеток у одноклеточных простейших паразитов». Паразиты и векторы. 4: 44. Дои:10.1186/1756-3305-4-44. ЧВК 3077326. PMID 21439063.
- ^ de Duve C (1996). «Рождение сложных клеток». Scientific American. 274 (4): 50–7. Bibcode:1996SciAm.274d..50D. Дои:10.1038 / scientificamerican0496-50. PMID 8907651.
- ^ Дьялл С.Д., Браун М.Т., Джонсон П.Дж. (2004). «Древние нашествия: от эндосимбионтов до органелл». Наука. 304 (5668): 253–7. Bibcode:2004Наука ... 304..253D. Дои:10.1126 / science.1094884. PMID 15073369. S2CID 19424594.
- ^ Кьяруги А., Московиц М.А. (2002). «Клеточная биология. PARP-1 - виновник апоптотической гибели клеток?». Наука. 297 (5579): 200–1. Дои:10.1126 / science.1074592. PMID 12114611. S2CID 82828773.
- ^ Качановски, С. Апоптоз: его происхождение, история, поддержание и медицинские последствия для рака и старения. Физ биол 13, http://iopscience.iop.org/article/10.1088/1478-3975/13/3/031001
- ^ а б c Шривастава, Ракеш (2007). Апоптоз, клеточная передача сигналов и болезни человека. Humana Press.