Ядерно-магнитная резонансная спектроскопия белков - Nuclear magnetic resonance spectroscopy of proteins
Ядерно-магнитная резонансная спектроскопия белков (обычно сокращенно белок ЯМР) - поле структурная биология в котором ЯМР-спектроскопия используется для получения информации о структуре и динамике белки, а также нуклеиновые кислоты, и их комплексы. Первым в этой области был Ричард Р. Эрнст и Курт Вютрих на ETH,[1] и по Ad Bax, Мариус Клор, Анжела Гроненборн на Национальные институты здравоохранения США,[2], и Герхард Вагнер в Гарвардский университет, среди прочего. Определение структуры методом ЯМР-спектроскопии обычно состоит из нескольких этапов, для каждой из которых используется отдельный набор узкоспециализированных методов. Подготавливается образец, проводятся измерения, применяются интерпретирующие подходы, рассчитывается и проверяется структура.
ЯМР включает квантово-механические свойства центрального ядра ("ядро ") атома. Эти свойства зависят от локального молекулярного окружения, и их измерение дает карту того, как атомы связаны химически, насколько близко они находятся в пространстве и как быстро они движутся относительно друг друга. Эти свойства в основном те же, что и в более знакомых магнитно-резонансная томография (МРТ), но в молекулярных приложениях используется несколько иной подход, соответствующий изменению масштаба с миллиметров (представляющих интерес для радиологов) на нанометры (связанные атомы обычно находятся на расстоянии доли нанометра друг от друга), то есть в миллион раз. Это изменение масштаба требует гораздо более высокой чувствительности обнаружения и стабильности для долгосрочных измерений. В отличие от МРТ, исследования структурной биологии не создают изображение напрямую, а полагаются на сложные компьютерные вычисления для создания трехмерных молекулярных моделей.
В настоящее время большинство образцов исследуют в растворе в воде, но методы разрабатываются также для работы с твердыми образцами. Сбор данных основан на помещении образца внутрь мощного магнита, передаче радиочастотных сигналов через образец и измерении поглощения этих сигналов. В зависимости от окружения атомов в белке ядра отдельных атомов будут поглощать различные частоты радиосигналов. Кроме того, сигналы поглощения разных ядер могут нарушаться соседними ядрами. Эта информация может быть использована для определения расстояния между ядрами. Эти расстояния, в свою очередь, можно использовать для определения общей структуры белка.
Типичное исследование может включать, как два белка взаимодействуют друг с другом, возможно, с целью разработки небольших молекул, которые можно использовать для исследования нормальной биологии взаимодействия ("химическая биология ") или предоставить возможные сведения о фармацевтическом использовании (разработка лекарств ). Часто взаимодействующая пара белков может быть идентифицирована исследованиями генетики человека, что указывает на то, что взаимодействие может быть нарушено неблагоприятными мутациями, или они могут играть ключевую роль в нормальной биологии «модельного» организма, такого как плодовая муха, дрожжи. , Червь C. elegans, или мышей. Для приготовления образца обычно используются методы молекулярной биологии, чтобы бактериальная ферментация. Это также позволяет изменить изотопный состав молекулы, что желательно, поскольку изотопы ведут себя по-разному и предоставляют методы для идентификации перекрывающихся сигналов ЯМР.
Базовые приготовления

Ядерный магнитный резонанс белков проводится на водных образцах с высоким содержанием очищенный белок. Обычно проба состоит из 300-600 микролитров с концентрацией белка от 0,1 до 3. миллимолярный. Источник белка может быть натуральным или производиться производственная система с помощью рекомбинантная ДНК методы через генная инженерия. Рекомбинантно выразил белки обычно легче производить в достаточном количестве, и этот метод делает изотопический маркировка возможный.
Очищенный белок обычно растворяют в буферный раствор и доводят до желаемых условий растворителя. Образец ЯМР готовят в тонкостенном стакане. трубка.
Сбор информации
ЯМР белков использует эксперименты с многомерным ядерным магнитным резонансом для получения информации о белке. В идеале каждое отдельное ядро в молекуле испытывает различное электронное окружение и, следовательно, имеет различную химический сдвиг по которому его можно узнать. Однако в больших молекулах, таких как белки, количество резонансов обычно может достигать нескольких тысяч, и одномерный спектр неизбежно имеет случайные перекрытия. Поэтому проводятся многомерные эксперименты, которые коррелируют частоты отдельных ядер. Дополнительные размеры уменьшают вероятность перекрытия и имеют большее информационное содержание, поскольку они коррелируют сигналы от ядер в определенной части молекулы. Намагничивание передается на образец с помощью импульсов электромагнитного (радиочастота ) энергии и между ядрами с использованием задержек; процесс описывается так называемыми последовательности импульсов. Последовательности импульсов позволяют экспериментатору исследовать и выбирать определенные типы связей между ядрами. Множество экспериментов по ядерному магнитному резонансу, используемых с белками, делятся на две основные категории: в одной намагниченность передается через химические связи, а во вторую - через пространство, независимо от структуры связывания. Первая категория используется для присвоения различных химические сдвиги к конкретному ядру, а второй в основном используется для создания ограничений расстояния, используемых при вычислении структуры и при назначении с немеченым белком.
В зависимости от концентрации образца, магнитного поля спектрометра и типа эксперимента, один эксперимент с многомерным ядерным магнитным резонансом на образце белка может занять часы или даже несколько дней, чтобы получить подходящее отношение сигнал / шум. посредством усреднения сигнала и для обеспечения достаточной эволюции передачи намагниченности в различных измерениях эксперимента. При прочих равных условиях эксперименты с более высокой размерностью займут больше времени, чем эксперименты с более низкой размерностью.
Как правило, первый эксперимент, который нужно измерить с меченным изотопом белком, представляет собой 2D гетероядерная одноквантовая корреляция (HSQC) спектр, где «гетероядерный» относится к ядрам, отличным от 1H. Теоретически гетероядерная одноквантовая корреляция имеет один пик для каждого H, связанного с гетероядром. Таким образом, в 15N-HSQC с 15N меченого белка, ожидается один сигнал для каждого атома азота в позвоночнике, за исключением пролин, который не имеет амид-водорода из-за циклической природы его основной цепи. Дополнительные сигналы 15N-HSQC вносятся каждым остатком с азотно-водородной связью в его боковой цепи (W, N, Q, R, H, K). 15N-HSQC часто называют «отпечатком пальца» белка, потому что каждый белок имеет уникальный паттерн сигнальных положений. Анализ 15N-HSQC позволяет исследователям оценить, присутствует ли ожидаемое количество пиков, и, таким образом, выявить возможные проблемы из-за множества конформации или неоднородность образца. Относительно быстрый эксперимент с одноквантовой гетероядерной корреляцией помогает определить возможность проведения последующих более длительных, более дорогих и сложных экспериментов. Невозможно приписать пики конкретным атомам только на основе одноканальной гетероядерной корреляции.
Назначение резонанса
Для анализа данных ядерного магнитного резонанса важно получить определение резонанса для белка, то есть выяснить, какие химический сдвиг соответствует какому атому. Обычно это достигается последовательная ходьба с использованием информации, полученной из нескольких различных типов экспериментов ЯМР. Точная процедура зависит от того, является ли белок изотопно меченый или нет, поскольку многие эксперименты по назначению зависят от углерода-13 и азота-15.
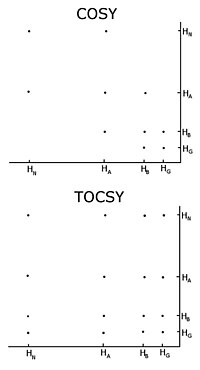
Гомоядерный ядерный магнитный резонанс
С немеченым белком обычная процедура состоит в том, чтобы записать серию двумерных экспериментов по гомоядерному магнитному резонансу с корреляционная спектроскопия (COSY), несколько типов которых включают обычную корреляционную спектроскопию, полная корреляция спектроскопия (TOCSY) и ядерный эффект Оверхаузера спектроскопия (NOESY).[3][4] Двумерный ядерный магнитный резонанс дает двумерный спектр. Единицами обеих осей являются химические сдвиги. COSY и TOCSY передают намагниченность через химические связи между соседними протонами. Обычный эксперимент корреляционной спектроскопии может передавать намагниченность только между протонами на соседних атомах, тогда как в эксперименте полной корреляционной спектроскопии протоны могут передавать намагниченность, поэтому она передается между всеми протонами, которые связаны соседними атомами. Таким образом, в обычной корреляционной спектроскопии альфа-протон передает намагниченность бета-протонам, бета-протоны передаются альфа- и гамма-протонам, если таковые имеются, затем гамма-протон передается бета- и дельта-протонам, и процесс продолжается. . В полной корреляционной спектроскопии альфа и все другие протоны могут передавать намагниченность в бета, гамма, дельта, эпсилон, если они связаны непрерывной цепочкой протонов. Непрерывная цепочка протонов - это боковая цепь индивидуума. аминокислоты. Таким образом, эти два эксперимента используются для построения так называемых спиновых систем, то есть составления списка резонансов химического сдвига пептидного протона, альфа-протонов и всех протонов от каждого из них. остаток Сайдчейн. Какие химические сдвиги соответствуют каким ядрам в спиновой системе, определяется обычными связями корреляционной спектроскопии и тем фактом, что разные типы протонов имеют характерные химические сдвиги. Чтобы соединить различные спиновые системы в последовательном порядке, необходимо использовать эксперимент ядерной спектроскопии эффекта Оверхаузера. Поскольку этот эксперимент передает намагниченность в пространстве, он покажет кросс-пики для всех протонов, находящихся близко в пространстве, независимо от того, находятся они в одной спиновой системе или нет. Соседние остатки по своей природе близки в пространстве, поэтому отнесения могут быть сделаны по пикам в NOESY с другими спиновыми системами.
Одной из важных проблем использования гомоядерного ядерного магнитного резонанса является перекрытие пиков. Это происходит, когда разные протоны имеют одинаковые или очень похожие химические сдвиги. Эта проблема усугубляется по мере того, как белок становится больше, поэтому гомоядерный ядерный магнитный резонанс обычно ограничивается небольшими белками или пептидами.
Ядерный магнитный резонанс Азот-15
Наиболее часто выполняемый эксперимент 15N - это 1ЧАС-15N HSQC. Эксперимент очень чувствителен и поэтому может быть проведен относительно быстро. Его часто используют для проверки пригодности белка для определения структуры с помощью ЯМР, а также для оптимизации условий образца. Это один из стандартных наборов экспериментов, используемых для определения структуры раствора белка. HSQC можно расширить до трехмерных и четырехмерных экспериментов ЯМР, таких как 15N-TOCSY-HSQC и 15N-NOESY-HSQC.[5]

Ядерный магнитный резонанс углерода-13 и азота-15
Когда белок помечен углеродом-13 и азотом-15, можно записывать эксперименты с тройным резонансом которые передают намагниченность по пептидной связи и, таким образом, связывают различные спиновые системы посредством связей.[6][7] Обычно это делается с помощью следующих экспериментов: HNCO, HN (CA) CO, HNCA,[8] HN (CO) CA, HNCACB и CBCA (CO) NH. Все шесть экспериментов состоят из 1ЧАС-15Плоскость N (похожая на спектр HSQC) расширена с размером углерода. В HN (CA) CO каждый HN Плоскость содержит пики карбонильного углерода из его остатка, а также предыдущий в последовательности. HNCO содержит химический сдвиг карбонильного углерода только от предыдущего остатка, но намного более чувствителен, чем HN (CA) CO. Эти эксперименты позволяют каждому 1ЧАС-15Пик N должен быть связан с предыдущим карбонильным углеродом, и затем может быть предпринято последовательное сопоставление путем сопоставления сдвигов каждого собственного и предыдущего углеродов спиновой системы. HNCA и HN (CO) CA работают аналогично, только с альфа-атомами углерода (Cα), а не карбонилы, а HNCACB и CBCA (CO) NH содержат как альфа-углерод, так и бета-углерод (Cβ). Обычно требуется несколько из этих экспериментов, чтобы устранить перекрытие в углеродном измерении. Эта процедура обычно менее неоднозначна, чем метод на основе NOESY, поскольку он основан на переводе облигаций. В методах, основанных на NOESY, появятся дополнительные пики, соответствующие атомам, которые расположены близко в пространстве, но не принадлежат последовательным остаткам, что затрудняет процесс присвоения. После первоначального последовательного назначения резонанса обычно можно расширить назначение от Cα и Cβ к остальной части боковой цепи с использованием таких экспериментов, как HCCH-TOCSY, который в основном является экспериментом TOCSY, разрешенным в дополнительном углеродном измерении.
Поколение сдержанности
Для проведения расчетов конструкции необходимо создать ряд экспериментально определенных ограничений. Они попадают в разные категории; наиболее широко используются ограничения расстояния и ограничения угла.
Ограничения расстояния
Кросспик в NOESY эксперимент означает пространственную близость между двумя рассматриваемыми ядрами. Таким образом, каждый пик можно преобразовать в максимальное расстояние между ядрами, обычно от 1,8 до 6 ангстремы. Интенсивность пика NOESY пропорциональна расстоянию до минус 6-й степени, поэтому расстояние определяется в соответствии с интенсивностью пика. Отношение интенсивности к расстоянию не является точным, поэтому обычно используется диапазон расстояний.
Очень важно отнести пики NOESY к правильным ядрам на основе химических сдвигов. Если эта задача выполняется вручную, это обычно очень трудоемко, поскольку белки обычно имеют тысячи пиков NOESY. Некоторые компьютерные программы, такие как PASD[9][10]/XPLOR-NIH,[11][12] UNIO,[13] ЦИАНА[14], ARIA[15]/ЦНС[16], и AUDANA[17]/PONDEROSA-C / S[18] в интегративной платформе ЯМР[19] выполнять эту задачу автоматически на предварительно обработанных вручную списках пиковых позиций и пиковых объемов, связанных с расчетом структуры. Прямой доступ к необработанным данным NOESY без громоздкой необходимости итеративно уточняемых списков пиков пока предоставляется только PASD.[10] алгоритм реализован в XPLOR-NIH[11], подход ATNOS / CANDID, реализованный в программном комплексе UNIO[13], и PONDEROSA-C / S, что действительно гарантирует объективный и эффективный спектральный анализ NOESY.
Для получения как можно более точных оценок большое преимущество имеет доступ к экспериментам NOESY по углероду-13 и азоту-15, поскольку они помогают устранить перекрытие в протонном измерении. Это приводит к более быстрым и надежным заданиям и, в свою очередь, к лучшим структурам.
Угловые ограничения
В дополнение к ограничению расстояния, ограничения на углы кручения химических связей, обычно углы psi и phi, могут быть созданы. Один из подходов - использовать Уравнение Карплюса, чтобы создать угловые ограничения из константы связи. Другой подход использует химические сдвиги для создания угловых ограничений. Оба метода используют тот факт, что геометрия вокруг альфа-углерода влияет на константы взаимодействия и химические сдвиги, поэтому, учитывая константы взаимодействия или химические сдвиги, можно сделать квалифицированное предположение о торсионных углах.
Ограничения ориентации
Молекулы аналита в образце можно частично упорядочить по отношению к внешнему магнитному полю спектрометра, манипулируя условиями образца. Общие методы включают добавление бактериофаги или же двуцеллы к образцу, или подготовка образца в растянутом полиакриламидный гель. Это создает локальную среду, которая способствует определенной ориентации несферических молекул. Обычно в растворе ЯМР диполярные связи между ядрами усредняются из-за быстрого переворачивания молекулы. Небольшая перенаселенность одной ориентации означает, что остаточная дипольная связь еще предстоит наблюдать. Диполярная связь обычно используется в твердотельный ЯМР и предоставляет информацию об относительной ориентации векторов связей относительно единой глобальной системы отсчета. Обычно ориентацию вектора N-H исследуют в эксперименте, подобном HSQC. Первоначально остаточные диполярные связи использовались для уточнения ранее определенных структур, но также были предприняты попытки определения структуры de novo.[20]
Водородно-дейтериевый обмен
ЯМР-спектроскопия специфична для ядра. Таким образом, он может различать водород и дейтерий. Протоны амида в белке легко обмениваются с растворителем, и, если растворитель содержит другой изотоп, обычно дейтерий, реакцию можно контролировать с помощью ЯМР-спектроскопии. Насколько быстро данный амид обменивается, отражает его доступность для растворителя. Таким образом, скорости обмена амида могут дать информацию о том, какие части белка погребены, связаны водородными связями и т. Д. Обычным применением является сравнение обмена в свободной форме с комплексной. Предполагается, что амиды, которые становятся защищенными в комплексе, находятся в интерфейсе взаимодействия.
Расчет конструкции
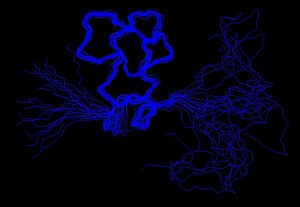
Установленные экспериментально ограничения могут использоваться в качестве входных данных для процесса расчета конструкции. Исследователи, использующие компьютерные программы, такие как XPLOR-NIH,[11] ЦИАНА или же GeNMR попытаться удовлетворить как можно больше ограничений в дополнение к общим свойствам белков, таким как длина связи и углы. Алгоритмы переводят ограничения и общие свойства белка в энергетические термины, а затем пытаются минимизировать эту энергию. Результатом этого процесса является ансамбль структур, которые, если бы данных было достаточно, чтобы определять определенную складку, сходились.
Проверка структуры
Важно отметить, что полученный ансамбль структур является «экспериментальной моделью», т. Е. Представлением определенного рода экспериментальных данных. Признание этого факта действительно важно, потому что это означает, что модель может быть хорошим или плохим представлением этих экспериментальных данных.[21] В общем, качество модели будет зависеть как от количества и качества экспериментальных данных, используемых для ее создания, так и от правильной интерпретации таких данных.
Важно помнить, что с каждым экспериментом связаны ошибки. Случайные ошибки повлияют на воспроизводимость и точность полученных конструкций. Если ошибки систематические, точность модели будут затронуты. Прецизионность указывает на степень воспроизводимости измерения и часто выражается как отклонение набора измеренных данных при тех же условиях. Однако точность указывает на степень приближения измерения к своему «истинному» значению.
В идеале модель белка будет тем точнее, чем больше соответствует действительная молекула, которая представляет, и будет более точной, поскольку имеется меньше неопределенности относительно положений их атомов. На практике не существует «стандартной молекулы», с которой можно было бы сравнивать модели белков, поэтому точность модели определяется степенью согласия между моделью и набором экспериментальных данных. Исторически структуры, определенные с помощью ЯМР, были, как правило, более низкого качества, чем структуры, определенные с помощью дифракции рентгеновских лучей. Частично это связано с меньшим количеством информации, содержащейся в данных, полученных с помощью ЯМР. Из-за этого факта стало обычной практикой определять качество ансамблей ЯМР, сравнивая его с уникальной конформацией, определенной с помощью дифракции рентгеновских лучей, для того же белка. Однако структура дифракции рентгеновских лучей может не существовать, и, поскольку белки в растворе представляют собой гибкие молекулы, белок, представленный единственной структурой, может привести к недооценке внутренней вариации атомных положений белка. Набор конформаций, определенный с помощью ЯМР или рентгеновской кристаллографии, может быть лучшим представлением экспериментальных данных белка, чем уникальная конформация.[22]
Полезность модели будет определяться, по крайней мере частично, степенью точности и точности модели. Точная модель с относительно низкой точностью может быть полезна для изучения эволюционных отношений между структурами набора белков, тогда как рациональный дизайн лекарств требует как точных, так и точных моделей. Модель, которая не является точной, независимо от степени точности, с которой она была получена, не будет очень полезной.[21][23]
Поскольку белковые структуры представляют собой экспериментальные модели, которые могут содержать ошибки, очень важно уметь обнаруживать эти ошибки. Процесс, направленный на обнаружение ошибок, известен как проверка. Существует несколько методов проверки структур, некоторые из которых являются статистическими, например ПРОЧЕК и ЧТО, ЕСЛИ в то время как другие основаны на физических принципах, как CheShift, или смесь статистических и физических принципов PSVS.
Динамика
Помимо конструкций, ядерный магнитный резонанс может дать информацию о динамике различных частей белок. Обычно это включает измерение времени релаксации, например T1 и т2 для определения параметров порядка, времен корреляции и скоростей химического обмена. Релаксация ЯМР является следствием локальных флуктуаций магнитные поля внутри молекулы. Локальные флуктуирующие магнитные поля создаются движением молекул. Таким образом, измерения времен релаксации могут предоставить информацию о движениях внутри молекулы на атомном уровне. В ЯМР-исследованиях динамики белков азот-15 Изотоп является предпочтительным ядром для изучения, потому что его времена релаксации относительно просто соотнести с молекулярными движениями. Однако это требует изотопного мечения белка. Т1 и т2 времена релаксации можно измерить с помощью различных типов HSQC эксперименты на основе. Типы движений, которые могут быть обнаружены, - это движения, которые происходят во времени в диапазоне от примерно 10 пикосекунд до примерно 10 наносекунд. Кроме того, можно изучать более медленные движения, которые имеют место в масштабе времени от 10 микросекунд до 100 миллисекунд. Однако, поскольку атомы азота находятся в основном в основе белка, результаты в основном отражают движения основы, которая является наиболее жесткой частью молекулы белка. Таким образом, результаты, полученные из азот-15 измерения релаксации могут не быть репрезентативными для всего белка. Следовательно, методы, использующие релаксационные измерения углерод-13 и дейтерий недавно были разработаны, что позволяет систематически изучать движения боковых цепей аминокислот в белках. Сложный и частный случай изучения динамики и гибкости пептидов и полноразмерных белков представлен неупорядоченными структурами. В настоящее время принято считать, что белки могут демонстрировать более гибкое поведение, известное как нарушение или отсутствие структуры; однако можно описать ансамбль структур вместо статической картины, представляющей полностью функциональное состояние белка. Многие достижения представлены в этой области, в частности, с точки зрения новых импульсных последовательностей, технологических усовершенствований и тщательной подготовки исследователей в этой области.
ЯМР-спектроскопия на больших белках
Традиционно спектроскопия ядерного магнитного резонанса ограничивалась относительно небольшими белками или белковыми доменами. Это частично вызвано проблемами с разрешением перекрывающихся пиков в более крупных белках, но это было облегчено введением изотопного мечения и многомерных экспериментов. Еще одна более серьезная проблема заключается в том, что в больших белках намагниченность релаксирует быстрее, а это означает, что у вас меньше времени для обнаружения сигнала. Это, в свою очередь, приводит к тому, что пики становятся шире и слабее и в конечном итоге исчезают. Были предложены два метода ослабления релаксации: оптимизированная для поперечной релаксации спектроскопия (TROSY)[24] и дейтерирование[25] белков. Используя эти методы, стало возможным изучать белки в комплексе с 900 кДа. сопровождающий GroES -GroEL.[26]
Автоматизация процесса
Определение структуры с помощью ЯМР традиционно было трудоемким процессом, требующим интерактивного анализа данных высококвалифицированным ученым. Был проявлен значительный интерес к автоматизации процесса, чтобы увеличить производительность определения структуры и сделать ЯМР белков доступным для неспециалистов (см. структурная геномика ). Двумя наиболее трудоемкими задействованными процессами являются назначение резонанса для конкретной последовательности (назначение основной и боковой цепи) и задачи назначения NOE. Было опубликовано несколько различных компьютерных программ, которые нацелены на отдельные части общего процесса определения структуры ЯМР в автоматическом режиме. Наибольший прогресс был достигнут в задаче автоматического присвоения NOE. До сих пор были предложены только подходы FLYA и UNIO для выполнения всего процесса определения структуры ЯМР белка в автоматическом режиме без какого-либо вмешательства человека.[13][14] В последнее время модули в NMRFAM-SPARKY например APES (двухбуквенный код: ae), I-PINE / PINE-SPARKY (двухбуквенный код: ep; Веб-сервер I-PINE ) и PONDEROSA (двухбуквенный код: c3, вверх; Веб-сервер PONDEROSA ) интегрированы таким образом, что обеспечивает полную автоматизацию с возможностью визуальной проверки на каждом этапе.[27] Также были предприняты усилия по стандартизации протокола расчета конструкции, чтобы сделать его более быстрым и поддающимся автоматизации.[28]
Смотрите также
- ЯМР-спектроскопия
- Ядерный магнитный резонанс
- Спектроскопия ядерного магнитного резонанса углеводов
- Спектроскопия ядерно-магнитного резонанса нуклеиновых кислот
- Кристаллизация белка
- Белковая динамика
- Релаксация (ЯМР)
- Рентгеновская кристаллография
Рекомендации
- ^ Вютрих К. (ноябрь 2001 г.). «Путь к ЯМР-структурам белков». Структурная и молекулярная биология природы. 8 (11): 923–5. Дои:10.1038 / nsb1101-923. PMID 11685234. S2CID 26153265.
- ^ Клор, Дж. Мариус (2011). «Приключения в биомолекулярном ЯМР» (PDF). В Харрис, Робин К.; Василишен, Родерик Л. (ред.). Энциклопедия магнитного резонанса. Джон Вили и сыновья. Дои:10.1002/9780470034590. HDL:11693/53364. ISBN 9780470034590.
- ^ Вютрих К. (декабрь 1990 г.). «Определение структуры белка в растворе методом ЯМР-спектроскопии». J. Biol. Chem. 265 (36): 22059–62. PMID 2266107.
- ^ Clore GM, Gronenborn AM (1989). «Определение трехмерных структур белков и нуклеиновых кислот в растворе». CRC Critical Reviews в биохимии и молекулярной биологии. 24 (5): 479–564. Дои:10.3109/10409238909086962. PMID 2676353.
- ^ Clore GM, Gronenborn AM (1991). «Структуры более крупных белков в растворе: трехмерная и четырехмерная гетероядерная спектроскопия ЯМР». Наука. 252 (5011): 1390–1399. Дои:10.1126 / science.2047852. OSTI 83376. PMID 2047852.
- ^ Clore GM, Gronenborn AM (1991). «Применение трехмерной и четырехмерной гетероядерной ЯМР-спектроскопии для определения структуры белков». Прогресс в спектроскопии ядерного магнитного резонанса. 23 (1): 43–92. Дои:10.1016 / 0079-6565 (91) 80002-Дж.
- ^ Bax A, Grzesiek S (1993). «Методические достижения в области ЯМР белков». Отчеты о химических исследованиях. 26 (4): 131–138. Дои:10.1021 / ar00028a001.
- ^ Bax A, Ikura M (май 1991 г.). «Эффективный метод 3D ЯМР для корреляции резонансов протона и амида основной цепи 15N с альфа-углеродом предыдущего остатка в белках, равномерно обогащенных 15N / 13C». J. Biomol. ЯМР. 1 (1): 99–104. Дои:10.1007 / BF01874573. PMID 1668719. S2CID 20037190.
- ^ Kuszewski J, Schwieters CD, Garrett DS, Byrd RA, Tjandra N, Clore GM (2004). «Полностью автоматизированное, очень устойчивое к ошибкам определение структуры макромолекул на основе многомерных ядерных спектров усиления Оверхаузера и назначений химического сдвига». Журнал Американского химического общества. 126 (20): 6258–6273. Дои:10.1021 / ja049786h. PMID 15149223.
- ^ а б Kuszewski J, Thottungal RA, Clore GM, Schwieters CD (2008). «Автоматическое определение устойчивой к ошибкам структуры макромолекул на основе многомерных ядерных спектров усиления Оверхаузера и назначений химического сдвига: повышенная надежность и производительность алгоритма PASD». Журнал биомолекулярного ЯМР. 41 (4): 221–239. Дои:10.1007 / s10858-008-9255-1. ЧВК 2575051. PMID 18668206.
- ^ а б c Schwieters CD; Кушевский JJ; Tjandra N; Clore GM (январь 2003 г.). «Пакет для определения молекулярной структуры Xplor-NIH ЯМР». J. Magn. Резон. 160 (1): 65–73. Bibcode:2003JMagR.160 ... 65S. Дои:10.1016 / S1090-7807 (02) 00014-9. PMID 12565051.
- ^ Schwieters, CD; Kuszewski, JJ; Клор, GM (2006). «Использование Xplor-NIH для определения молекулярной структуры ЯМР». Прогресс в спектроскопии ядерного магнитного резонанса. 48 (1): 47–62. Дои:10.1016 / j.pnmrs.2005.10.001.
- ^ а б c Херрманн Т. (2010). «Расчет структуры белка и автоматизированные ограничения NOE». Энциклопедия магнитного резонанса. Дои:10.1002 / 9780470034590.emrstm1151. ISBN 978-0470034590.
- ^ а б Гюнтерт П. (2004). «Автоматизированный расчет структуры ЯМР с помощью CYANA». Методы ЯМР белков. Методы Мол. Биол. 278. С. 353–78. CiteSeerX 10.1.1.332.4843. Дои:10.1385/1-59259-809-9:353. ISBN 978-1-59259-809-0. PMID 15318003.
- ^ Рипинг W; Habeck M; Bardiaux B; Бернар А; Маллявин TE; Нилгес М (февраль 2007 г.). «ARIA2: автоматическое определение NOE и интеграция данных в расчет структуры ЯМР». Биоинформатика. 23 (3): 381–2. Дои:10.1093 / биоинформатика / btl589. PMID 17121777.
- ^ Брюнгер, АТ; Адамс, PD; Clore, GM; ДеЛано, Вашингтон; Gros, P; Гросс-Кунстлеве, RW; Цзян, JS; Кушевский, Дж; Nilges, M; Pannu, NS; Читать, RJ; Рис, LM; Саймонсон, Т; Уоррен, Г.Л. (1 сентября 1998 г.). «Система кристаллографии и ЯМР: новый пакет программ для определения структуры макромолекул». Acta Crystallographica Раздел D. 54 (Pt 5): 905–21. Дои:10.1107 / s0907444998003254. PMID 9757107.
- ^ Ли В., Пети К.М., Корнилеску Дж., Старк Дж. Л., Маркли Дж. Л. (июнь 2016 г.). «Алгоритм AUDANA для автоматического определения трехмерной структуры белка по данным ЯМР NOE». J. Biomol. ЯМР. 65 (2): 51–7. Дои:10.1007 / s10858-016-0036-у. ISSN 0925-2738. ЧВК 4921114. PMID 27169728.
- ^ Ли, Вунхи; Старк, Хайме Л .; Маркли, Джон Л. (2014-11-01). «PONDEROSA-C / S: программный комплекс на базе клиент-сервер для автоматизированного определения трехмерной структуры белков». Журнал биомолекулярного ЯМР. 60 (2–3): 73–75. Дои:10.1007 / s10858-014-9855-х. ISSN 0925-2738. ЧВК 4207954. PMID 25190042.
- ^ Ли, Вунхи; Корнилеску, Габриэль; Дашти, Хесам; Eghbalnia, Hamid R .; Тонелли, Марко; Вестлер, Уильям М .; Мясник, Сэмюэл Э .; Henzler-Wildman, Katherine A .; Маркли, Джон Л. (2016-04-01). «Интегративный ЯМР для биомолекулярных исследований». Журнал биомолекулярного ЯМР. 64 (4): 307–332. Дои:10.1007 / s10858-016-0029-х. ISSN 0925-2738. ЧВК 4861749. PMID 27023095.
- ^ де Альба Э; Тяндра Н (2004). Остаточные диполярные связи в определении структуры белка. Методы Мол. Биол. 278. С. 89–106. Дои:10.1385/1-59259-809-9:089. ISBN 978-1-59259-809-0. PMID 15317993.
- ^ а б Ласковски, Р. А. (2003). «Структурное обеспечение качества». Структурная биоинформатика. Методы биохимического анализа. 44. С. 273–303. Дои:10.1002 / 0471721204.ch14. ISBN 9780471202004. PMID 12647391.
- ^ Арнаутова, Ю. А .; Vila, J. A .; Мартин, О. А .; Шерага, Х.А. (2009). «Что мы можем узнать, вычислив химические сдвиги 13Calpha для рентгеновских моделей белков?». Acta Crystallographica Раздел D. 65 (7): 697–703. Дои:10.1107 / S0907444909012086. ЧВК 2703576. PMID 19564690.
- ^ Spronk, C.A .; Набуурс, С.Б .; Krieger, E .; Vriend, G .; Вуистер, Г. В. (2004). «Проверка белковых структур, полученных с помощью ЯМР-спектроскопии». Прогресс в спектроскопии ядерного магнитного резонанса. 45 (3–4): 315–337. Дои:10.1016 / j.pnmrs.2004.08.003.
- ^ Первушин К; Riek R; Более широкий G; Вютрих К. (ноябрь 1997 г.). «Ослабленная релаксация T2 за счет взаимного подавления диполь-дипольного взаимодействия и анизотропии химического сдвига указывает на возможность структурного ЯМР очень больших биологических макромолекул в растворе». Proc. Natl. Акад. Sci. СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ. 94 (23): 12366–71. Bibcode:1997PNAS ... 9412366P. Дои:10.1073 / пнас.94.23.12366. ЧВК 24947. PMID 9356455.
- ^ Маркус М.А.; Dayie KT; Matsudaira P; Вагнер Г. (октябрь 1994 г.). «Влияние дейтерирования на скорость релаксации амидных протонов в белках. Гетероядерные эксперименты ЯМР на виллине 14T». J Магн Резон B. 105 (2): 192–5. Bibcode:1994JMRB..105..192M. Дои:10.1006 / jmrb.1994.1122. PMID 7952934.
- ^ Fiaux J; Bertelsen EB; Хорвич А.Л .; Вютрих К. (июль 2002 г.). «ЯМР-анализ комплекса 900K GroEL GroES». Природа. 418 (6894): 207–11. Дои:10.1038 / природа00860. PMID 12110894. S2CID 2451574.
- ^ Ли, Вунхи; Тонелли, Марко; Маркли, Джон Л. (2015-04-15). «NMRFAM-SPARKY: усовершенствованное программное обеспечение для биомолекулярной ЯМР-спектроскопии». Биоинформатика. 31 (8): 1325–1327. Дои:10.1093 / биоинформатика / btu830. ISSN 1367-4803. ЧВК 4393527. PMID 25505092.
- ^ Лю Г, Шэнь Й; Атрея HS; и другие. (Июль 2005 г.). «Протокол сбора и анализа данных ЯМР для высокопроизводительного определения структуры белка». Proc. Natl. Акад. Sci. СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ. 102 (30): 10487–92. Bibcode:2005PNAS..10210487L. Дои:10.1073 / pnas.0504338102. ЧВК 1180791. PMID 16027363.
дальнейшее чтение
- Т. Кевин Хитченс; Гордон С. Рул (2005). Основы ЯМР-спектроскопии белков (основное внимание уделяется структурной биологии). Берлин: Springer. ISBN 978-1-4020-3499-2.
- Куинси Тэн (2005). Структурная биология: практическое применение ЯМР. Берлин: Springer. Bibcode:2005stbi.book ..... т. ISBN 978-0-387-24367-2.
- Марк Рэнс; Кавана, Джон; Уэйн Дж. Фэйрбразер; Артур В. Хант III; Скелтон, Николас Дж. (2007). ЯМР-спектроскопия белков: принципы и практика (2-е изд.). Бостон: Academic Press. ISBN 978-0-12-164491-8.
- Курт Вютрих (1986). ЯМР белков и нуклеиновых кислот. Нью-Йорк: Вили. ISBN 978-0-471-82893-8.
внешняя ссылка
| Библиотечные ресурсы о Ядерно-магнитная резонансная спектроскопия белков |
- Стратегия на основе NOESY для назначения резонансов основной и боковой цепи больших белков без дейтерирования (протокол)
- расслабляться Программное обеспечение для анализа динамики ЯМР
- ProSA-web Веб-сервис для распознавания ошибок в экспериментально или теоретически определенных структурах белков
- Определение структуры белка по немногочисленным экспериментальным данным - вводная презентация
- ЯМР белков Белковые ЯМР эксперименты