Аргининосукцинатлиаза - Argininosuccinate lyase
| Аргининосукцинатлиаза | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 Кристаллическая структура аргининосукцинатлиазы утки со связанным аргининосукцинатом.[1] | |||||||||
| Идентификаторы | |||||||||
| Номер ЕС | 4.3.2.1 | ||||||||
| Количество CAS | 9027-34-3 | ||||||||
| Базы данных | |||||||||
| IntEnz | Просмотр IntEnz | ||||||||
| БРЕНДА | BRENDA запись | ||||||||
| ExPASy | Просмотр NiceZyme | ||||||||
| КЕГГ | Запись в KEGG | ||||||||
| MetaCyc | метаболический путь | ||||||||
| ПРИАМ | профиль | ||||||||
| PDB структуры | RCSB PDB PDBe PDBsum | ||||||||
| Генная онтология | AmiGO / QuickGO | ||||||||
| |||||||||
| Аргининосукцинатлиаза | |||||||
|---|---|---|---|---|---|---|---|
 Кристаллографическая структура мономера ASL человека с мечеными доменами.[2] | |||||||
| Идентификаторы | |||||||
| Символ | ASL | ||||||
| Ген NCBI | 435 | ||||||
| HGNC | 746 | ||||||
| OMIM | 608310 | ||||||
| RefSeq | NM_000048 | ||||||
| UniProt | P04424 | ||||||
| Прочие данные | |||||||
| Номер ЕС | 4.3.2.1 | ||||||
| Locus | Chr. 7 pter-q22 | ||||||
| |||||||
ASL (аргининосукцинатлиаза, также известный как аргининосукциназа) является фермент что катализирует обратимый распад аргининосукцинат (ASA) производит аминокислоту аргинин и дикарбоновая кислота фумарат. ASL, расположенный в цитозоле печени, является четвертым ферментом цикл мочевины и участвует в биосинтезе аргинина у всех видов и производстве мочевины в уреотелический разновидность.[2] Мутации в ASL, приводящие к низкой активности фермента, повышают уровень мочевины в организме и приводят к различным побочным эффектам.
Ген ASL расположен на хромосома 7 между центромера (соединение длинного и короткого плеча) и длинного (q) плеча в позиции 11.2, от базовая пара 64 984 963 на базовую пару 65 002 090.
ASL связан с внутригенная комплементация.[3][4][5]
Структура
ASL состоит из четырех идентичных мономеров; каждый мономер, состоящий из одной полипептидной цепи от 49 до 52 кДа,[6] от 196 до 208 кДа для всего тетрамерного фермента. Каждый мономер имеет три высококонсервативных участка, удаленных друг от друга, но эти участки группируются вместе в тетрамере, образуя четыре активных центра. Следовательно, каждый гомотетрамер ASL имеет четыре активных центра, катализирующих распад аргининосукцината.
Каждый мономер в гомотетрамере ASL состоит из трех структурных доменов; все три в основном альфа-спиральные. Домены 1 и 3 похожи по структуре, поскольку оба они состоят из мотивов спираль-поворот-спираль. Домен 1 мономера содержит амино-конец. Домен 2 содержит один небольшой бета-лист, девять альфа-спиралей и карбоксильный конец. Три из девяти альфа-спиралей на одном мономере участвуют в основном в гидрофобных взаимодействиях с другим мономером с образованием димера. Затем два димера соединяются посредством альфа-спирали, по одному от каждого мономера, с образованием центрального ядра из 20 спиралей. Объединение всех четырех мономеров обеспечивает каталитическую активность в каждом возможном активном центре.[4]
Внутригенное дополнение
Множественные копии полипептида, кодируемого ген часто может образовывать агрегат, называемый мультимером. Когда мультимер образуется из полипептидов, продуцируемых двумя разными мутант аллели конкретного гена смешанный мультимер может проявлять большую функциональную активность, чем несмешанные мультимеры, образованные каждым из мутантов по отдельности. Когда смешанный мультимер демонстрирует повышенную функциональность по сравнению с несмешанными мультимерами, это явление упоминается как внутригенная комплементация. У человека ASL представляет собой мультимерный (тетрамерный) белок. Расстройство ASL у людей может возникать в результате мутаций в гене ASL, особенно мутаций, которые влияют на активный сайт мутантного мультимерного белка. Расстройство ASL связано со значительной клинической и генетической гетерогенностью, которая, как считается, отражает обширную внутригенную комплементацию, имеющую место у отдельных пациентов.[3][4][5]
Механизм
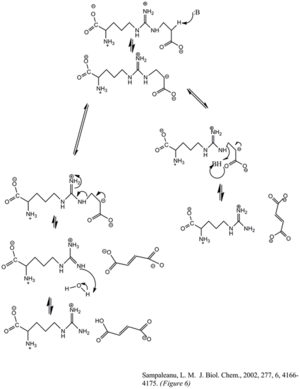

Расщепление ферментом аргининосукцината с образованием фумарата и аргинина происходит посредством реакции элиминирования E1cb. Основание инициирует реакцию, депротонируя углерод, прилегающий к аргинину, или уходящую группу. Недавние мутагенные исследования гомологов ASL показали, что гистидин 162 или треонин 161 из ASL отвечает за отрыв протонов от Cβ, прямо или косвенно через молекулу воды.[6] Полагают, что лизин 289 стабилизирует отрицательно заряженный промежуточный карбанион. Хотя нет единого мнения о каталитической кислоте, которая отдает протон иминной функциональной группе аргининового продукта, некоторые исследования мутагенеза показывают, что в этом может участвовать серин 283.[6]
Роль в цикле мочевины
Аммиак (NH3) является токсичным веществом для многих аэробных организмов и должен выводиться из организма. Некоторые водные организмы выделяют токсин прямо в окружающую среду, в то время как другие уреотелические виды должны преобразовывать свои токсичные азотные отходы в нетоксичные компоненты, такие как мочевая кислота или мочевина, с помощью серии катализируемых этапов, более известных как цикл мочевины. ASL катализирует четвертый этап цикла, следующий за действием аргининосукцинатсинтетазы (ASS) в цитозоле печени. В то время как ASS катализирует образование аргининосукцината из цитруллина и аспартата, ASL расщепляет вновь образованный аргининосукцинат на L-аргинин и фумарат. L-аргинин продолжает цикл мочевины с образованием мочевины и орнитина, в то время как фумарат может входить в цикл лимонной кислоты.[7]
дельта-кристаллин
ASL, δ-кристаллин, фумараза класса II, аспартаза, аденилосукциназалиаза и 3-карбоксицис- и цис-муконат лактонизирующий фермент являются членами одного и того же гомотетрамерного суперсемейства ферментов, в которых большинство из них катализируют реакции элиминирования одного и того же типа, в которых присутствует связь CO или CN. сломанный и фумарат выпускается как продукт. δ-кристаллины являются основными структурными водорастворимыми белками хрусталика глаза большинства птиц, рептилий и некоторых других позвоночных.[4]
Внутри суперсемейства ASL наиболее тесно связан с δ-кристаллином по аминокислотной последовательности и складчатой структуре белка. Существует две изоформы δ-кристаллина, δI и δII. Эти две изоформы сохраняют 69% и 71% аминокислотной последовательности ASL, соответственно, но только изоформа δII сохраняет ту же ферментативную активность, что и ASL. Сходство привело исследователей к мысли, что эти кристаллины эволюционировали от привлечения к линзе уже существующих метаболических ферментов, таких как ASL, в процессе, называемом «совместное использование генов». Один и тот же генный продукт действует как кристаллин хрусталика и как фермент в других неокулярных тканях. Сравнительные исследования δ-кристаллинов были полезны для понимания ферментативного механизма реакции ASL.[8]
Мутации и дефицит ASL: аргинино-янтарная ацидурия
Мутации в гене ASL человека вызывают аргинино-янтарную ацидурию, редкое аутосомно-рецессивное заболевание, и приводят к нарушениям цикла мочевины. Аргининосукцинатлиаза - это промежуточный фермент в пути синтеза мочевины, и его функция необходима для продолжения цикла. Неработающий фермент приводит к накоплению у пациентов аммиака, аргининосукцината и цитруллина в крови, а аргининосукцинат выводится с мочой.[9] Другие результирующие симптомы включают летаргию, рвоту, переохлаждение, гипервентиляцию, гепатомегалию и прогрессирующую энцефалопатию у новорожденных, а также аномальный рост волос, фиброз печени, эпизодическую рвоту, задержку роста и развития.[9] у пациентов, которые позже в детстве страдают этим расстройством.
ASL является ключевым ферментом в превращении аммиака в мочевину в цикле мочевины. Уровень аммиака становится токсичным, что приводит к гипераммониемии.[10] Аммиак токсичен отчасти потому, что влияет на нервную систему. Существуют биохимические доказательства того, что повышение уровня аммиака может ингибировать глутаминазу и, следовательно, ограничивать скорость синтеза нейромедиаторов, таких как глутамат,[11] что может объяснить задержку развития у пациентов с аргинино-янтарной ацидурией.
Одна мутация у пациентов с аргинино-янтарной ацидурией возникает, когда глутамин 286 превращается в аргинин. Фермент теперь имеет положительно заряженный аргинин вместо нейтрально заряженного глутамина, и исследования показывают, что это изменение может стерически и / или электростатически препятствовать конформационным изменениям, необходимым для катализа.
Рекомендации
- ^ PDB: 1TJW; Сампалеану Л.М., Коддинг П.В., Лобсанов Ю.Д., Цай М., Смит Г.Д., Хорватин С., Хауэлл П.Л. (декабрь 2004 г.). «Структурные исследования мутантов дельта2 кристаллина утки дают представление о роли Thr161 и петли 280s в катализе». Biochem. J. 384 (Pt 2): 437–47. Дои:10.1042 / BJ20040656. ЧВК 1134128. PMID 15320872.
- ^ а б PDB: 1К62; Сампалеану Л.М., Валле Ф., Томпсон Г.Д., Хауэлл П.Л. (декабрь 2001 г.). «Трехмерная структура аргининосукцинатлиазы, часто дополняющей аллель Q286R». Биохимия. 40 (51): 15570–80. Дои:10.1021 / bi011525m. PMID 11747432.
- ^ а б Тернер М.А., Симпсон А., Макиннес Р.Р., Хауэлл П.Л. (август 1997 г.). «Аргининосукцинатлиаза человека: структурная основа для внутригенной комплементации». Proc. Natl. Акад. Sci. СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ. 94 (17): 9063–8. Дои:10.1073 / пнас.94.17.9063. ЧВК 23030. PMID 9256435.
- ^ а б c d Ю. Б., Хауэлл П. Л. (октябрь 2000 г.). «Внутригенная комплементация, структура и функция аргининосукцинатлиазы». Клетка. Мол. Life Sci. 57 (11): 1637–51. Дои:10.1007 / PL00000646. PMID 11092456. S2CID 1254964.
- ^ а б Ю Б., Томпсон Г. Д., Ип П., Хауэлл П. Л., Дэвидсон А. Р. (декабрь 2001 г.). «Механизмы внутригенной комплементации в локусе аргининосукцинатлиазы человека». Биохимия. 40 (51): 15581–90. Дои:10.1021 / bi011526e. PMID 11747433.
- ^ а б c Сампалеану Л. М., Ю. Б., Хауэлл П. Л. (февраль 2002 г.). «Мутационный анализ дельта-2-кристаллина утки и структура неактивного мутанта со связанным субстратом позволяют понять ферментативный механизм аргининосукцинатлиазы». J. Biol. Chem. 277 (6): 4166–75. Дои:10.1074 / jbc.M107465200. PMID 11698398.
- ^ Пратт, Шарлотта Амерли; Воет, Дональд; Воет, Джудит Г. (2008). «Рисунок 20.8». Основы биохимии: жизнь на молекулярном уровне. Нью-Йорк: Вили. ISBN 978-0-470-12930-2.
- ^ Чакраборти А.Р., Дэвидсон А., Хауэлл П.Л. (февраль 1999 г.). «Мутационный анализ аминокислотных остатков, участвующих в аргининосукцинатлиазной активности в кристаллине дельта II утки». Биохимия. 38 (8): 2435–43. Дои:10.1021 / bi982150g. PMID 10029537.
- ^ а б Фичичиоглу К., Манделл Р., Ши В.Е. (ноябрь 2009 г.). «Дефицит аргининосукцинатлиазы: отдаленные результаты у 13 пациентов, выявленные при скрининге новорожденных». Мол. Genet. Метаб. 98 (3): 273–7. Дои:10.1016 / j.ymgme.2009.06.011. ЧВК 2773214. PMID 19635676.
- ^ «Аргининосукцинатлиаза гена ASL». Национальные институты здравоохранения США. Министерство здравоохранения и социальных служб США. 2007 г.
- ^ Джек, JJB (1982). «Воздействие аммиака на центральную нервную систему». Журнал наследственных метаболических заболеваний. 5 (S2): 104. Дои:10.1007 / BF01805572. S2CID 33915515.