Фермент - Enzyme

| Часть серии по |
| Биохимия |
|---|
 |
| Ключевые компоненты |
| История биохимии |
| Глоссарии |
| Порталы: Биохимия |
Ферменты /ˈɛпzаɪмz/ находятся белки которые действуют как биологический катализаторы (биокатализаторы). Катализаторы ускоряют химические реакции. Молекулы, на которые могут действовать ферменты, называются субстраты, а фермент превращает субстраты в различные молекулы, известные как товары. Почти все метаболические процессы в клетка необходимость ферментативный катализ чтобы происходить с достаточно высокой скоростью, чтобы поддерживать жизнь.[1]:8.1 Метаболические пути зависят от ферментов, которые катализируют отдельные стадии. Исследование ферментов называется энзимология и новое поле псевдоферментный анализ недавно вырос, осознав, что в ходе эволюции некоторые ферменты утратили способность проводить биологический катализ, что часто отражается в их аминокислота последовательности и необычные «псевдокаталитические» свойства.[2][3]
Известно, что ферменты катализируют более 5000 типов биохимических реакций.[4] Другие биокатализаторы: каталитические молекулы РНК, называемые рибозимами. Ферменты ' специфичность происходит из их уникальных трехмерные конструкции.
Как и все катализаторы, ферменты увеличивают скорость реакции снижая ее энергия активации. Некоторые ферменты могут ускорить превращение субстрата в продукт во много миллионов раз. Крайний пример - оротидин-5'-фосфатдекарбоксилаза, что позволяет за миллисекунды протекать реакции, которая в противном случае заняла бы миллионы лет.[5][6] В химическом отношении ферменты подобны любому катализатору, они не расходуются в химических реакциях и не изменяют равновесие реакции. Ферменты отличаются от большинства других катализаторов гораздо более специфичными. На активность фермента могут влиять другие молекулы: ингибиторы представляют собой молекулы, которые снижают активность ферментов, и активаторы представляют собой молекулы, повышающие активность. Многие терапевтические наркотики и яды являются ингибиторами ферментов. Активность фермента заметно снижается за пределы оптимальной температура и pH, и многие ферменты (навсегда) денатурированный при чрезмерном нагревании теряет свою структуру и каталитические свойства.
Некоторые ферменты используются в коммерческих целях, например, при синтезе антибиотики. В некоторых товарах для дома используются ферменты для ускорения химических реакций: ферменты в биологических стиральные порошки расщеплять белок, крахмал или толстый пятна на одежде и ферменты в мясорубка расщепляют белки на более мелкие молекулы, благодаря чему мясо легче жевать.
Этимология и история

К концу 17 - началу 18 вв. Переваривание мясо выделениями желудка[7] и преобразование крахмал к сахара экстрактами растений и слюна были известны, но механизмы, с помощью которых это произошло, не были идентифицированы.[8]
Французский химик Ансельм Пайен был первым, кто открыл фермент, диастаза, в 1833 г.[9] Спустя несколько десятилетий при изучении ферментация сахара в алкоголь к дрожжи, Луи Пастер пришел к выводу, что это брожение было вызвано жизненная сила содержащиеся в дрожжевых клетках, называемые «ферментами», которые, как считалось, функционируют только в живых организмах. Он писал, что «алкогольное брожение - это действие, связанное с жизнью и организацией дрожжевых клеток, а не со смертью или разложением клеток».[10]
В 1877 году немецкий физиолог Вильгельм Кюне (1837–1900) впервые использовал термин фермент, который исходит из Греческий ἔνζυμον, «дрожжевой» или «дрожжевой», чтобы описать этот процесс.[11] Слово фермент позже использовался для обозначения неживых веществ, таких как пепсин, и слово брожение был использован для обозначения химической активности, производимой живыми организмами.[12]
Эдуард Бюхнер представил свою первую работу по изучению дрожжевых экстрактов в 1897 году. В серии экспериментов на Берлинский университет, он обнаружил, что сахар ферментировался дрожжевыми экстрактами, даже когда в смеси не было живых дрожжевых клеток.[13] Он назвал фермент, вызывающий ферментацию сахарозы "зимаза ".[14] В 1907 году он получил Нобелевская премия по химии за «открытие бесклеточной ферментации». Следуя примеру Бюхнера, ферменты обычно называют в зависимости от реакции, которую они проводят: суффикс -ase сочетается с названием субстрат (например., лактаза это фермент, который расщепляет лактоза ) или типу реакции (например, ДНК-полимераза образует полимеры ДНК).[15]
В начале 1900-х годов биохимическая идентичность ферментов была еще неизвестна. Многие ученые заметили, что ферментативная активность связана с белками, но другие (например, лауреат Нобелевской премии) Ричард Вильштеттер ) утверждал, что белки были просто носителями настоящих ферментов и что белки как таковой были неспособны к катализу.[16] В 1926 г. Джеймс Б. Самнер показали, что фермент уреаза был чистым белком и кристаллизовал его; он сделал то же самое для фермента каталаза в 1937 г. Заключение о том, что чистые белки могут быть ферментами, было окончательно продемонстрировано Джон Ховард Нортроп и Венделл Мередит Стэнли, который работал над пищеварительными ферментами пепсин (1930), трипсин и химотрипсин. Эти трое ученых были удостоены Нобелевской премии по химии 1946 года.[17]
Открытие того, что ферменты могут быть кристаллизованы, в конечном итоге позволило решить их структуру с помощью рентгеновская кристаллография. Впервые это было сделано для лизоцим, фермент, содержащийся в слезах, слюне и яичные белки который переваривает оболочку некоторых бактерий; структура была решена группой во главе с Дэвид Чилтон Филлипс и опубликовано в 1965 году.[18] Эта структура лизоцима с высоким разрешением положила начало области структурная биология и попытка понять, как работают ферменты, на атомарном уровне детализации.[19]
Соглашения об именах
Название фермента часто происходит от его субстрата или химической реакции, которую он катализирует, причем слово заканчивается на -ase.[1]:8.1.3 Примеры лактаза, алкогольдегидрогеназа и ДНК-полимераза. Различные ферменты, катализирующие одну и ту же химическую реакцию, называются изоферменты.[1]:10.3
В Международный союз биохимии и молекулярной биологии разработали номенклатура для ферментов Номера ЕС; Каждый фермент описывается последовательностью из четырех чисел, перед которой стоит «ЕС», что означает «Комиссия по ферментам». Первое число широко классифицирует фермент на основе его механизма.[20]
Классификация верхнего уровня:
- EC 1, Оксидоредуктазы: катализировать окисление / восстановительные реакции
- EC 2, Трансферазы: передать функциональная группа (например метильная или фосфатная группа)
- EC 3, Гидролазы: катализировать гидролиз различных облигаций
- EC 4, Лиасы: расщепляет различные связи способами, отличными от гидролиза и окисления
- EC 5, Изомеразы: катализировать изомеризация изменения в одной молекуле
- EC 6, Лигазы: соединить две молекулы с ковалентные связи.
Эти разделы подразделяются на другие элементы, такие как подложка, продукты и химический механизм. Фермент полностью определяется четырьмя числовыми обозначениями. Например, гексокиназа (EC 2.7.1.1) представляет собой трансферазу (EC 2), которая добавляет фосфатную группу (EC 2.7) к гексозному сахару, молекуле, содержащей спиртовую группу (EC 2.7.1).[21]
Структура

Ферменты обычно глобулярные белки, действуя самостоятельно или в больших комплексы. Последовательность аминокислот определяет структуру, которая, в свою очередь, определяет каталитическую активность фермента.[22] Хотя структура определяет функцию, новую ферментативную активность еще нельзя предсказать, основываясь только на структуре.[23] Структуры ферментов разворачиваются (денатурировать ) при нагревании или воздействии химических денатурирующих веществ, и это нарушение структуры обычно вызывает потерю активности.[24] Денатурация ферментов обычно связана с температурами выше нормального уровня вида; в результате ферменты бактерий, живущих в вулканических средах, таких как горячие источники ценятся промышленными пользователями за их способность функционировать при высоких температурах, что позволяет проводить реакции, катализируемые ферментами, с очень высокой скоростью.
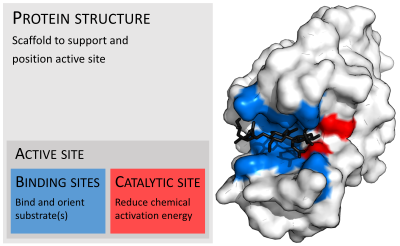
Ферменты обычно намного больше, чем их субстраты. Размеры варьируются от 62 аминокислотных остатков для мономер из 4-оксалокротонат таутомераза,[25] до более 2500 остатков в животном синтаза жирных кислот.[26] Лишь небольшая часть их структуры (около 2–4 аминокислот) непосредственно участвует в катализе: каталитический центр.[27] Этот каталитический сайт расположен рядом с одним или несколькими участок связывания где остатки ориентируют подложки. Каталитический сайт и сайт связывания вместе составляют фермент активный сайт. Оставшаяся большая часть структуры фермента служит для поддержания точной ориентации и динамики активного центра.[28]
В некоторых ферментах аминокислоты не принимают непосредственного участия в катализе; вместо этого фермент содержит сайты для связывания и ориентации каталитических кофакторы.[28] Ферментные структуры также могут содержать аллостерические сайты где связывание небольшой молекулы вызывает конформационное изменение что увеличивает или уменьшает активность.[29]
Небольшое количество РНК биологические катализаторы, называемые рибозимы существуют, которые снова могут действовать самостоятельно или в комплексе с белками. Наиболее распространенным из них является рибосома который представляет собой комплекс белковых и каталитических компонентов РНК.[1]:2.2
Механизм
Привязка субстрата
Ферменты должны связать свои субстраты, прежде чем они смогут катализировать какую-либо химическую реакцию. Ферменты обычно очень специфичны в отношении того, что субстраты они связываются, а затем катализируется химическая реакция. Специфика достигается за счет связывания карманов дополнительной формы, заряда и гидрофильный /гидрофобный характеристики к субстратам. Таким образом, ферменты могут различать очень похожие молекулы субстрата. хемоселективный, региоселективный и стереоспецифический.[30]
Некоторые из ферментов, показывающих самую высокую специфичность и точность, участвуют в копировании и выражение из геном. Некоторые из этих ферментов имеют "корректура "механизмы. Здесь фермент, такой как ДНК-полимераза катализирует реакцию на первом этапе, а затем проверяет соответствие продукта на втором этапе.[31] Этот двухэтапный процесс приводит к средней частоте ошибок менее 1 ошибки на 100 миллионов реакций в высокоточных полимеразах млекопитающих.[1]:5.3.1 Подобные механизмы корректуры также встречаются в РНК-полимераза,[32] аминоацил тРНК синтетазы[33] и рибосомы.[34]
И наоборот, некоторые ферменты ферментная неразборчивость, обладающие широкой специфичностью и действующие на ряд различных физиологически значимых субстратов. Многие ферменты обладают небольшой побочной активностью, которая возникла случайно (т. Е. нейтрально ), что может стать отправной точкой для эволюционного выбора новой функции.[35][36]
Модель "Замок и ключ"
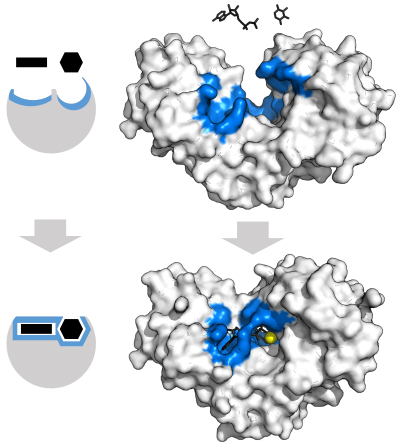
Чтобы объяснить наблюдаемую специфичность ферментов, в 1894 г. Эмиль Фишер предположили, что и фермент, и субстрат имеют определенные дополнительные геометрические формы, которые точно соответствуют друг другу.[37] Это часто называют моделью «замок и ключ».[1]:8.3.2 Эта ранняя модель объясняет специфичность ферментов, но не может объяснить стабилизацию переходного состояния, достигаемого ферментами.[38]
Модель индуцированной подгонки
В 1958 г. Даниэль Кошланд предложили модификацию модели «замок и ключ»: поскольку ферменты представляют собой довольно гибкие структуры, активный центр постоянно меняет форму в результате взаимодействий с субстратом, поскольку субстрат взаимодействует с ферментом.[39] В результате субстрат не просто связывается с жестким активным сайтом; аминокислота боковые цепи которые составляют активный центр, сформованы в точных положениях, которые позволяют ферменту выполнять свою каталитическую функцию. В некоторых случаях, например, гликозидазы, субстрат молекула также немного меняет форму при входе в активный сайт.[40] Активный центр продолжает изменяться до тех пор, пока субстрат не будет полностью связан, после чего определяется окончательная форма и распределение заряда.[41]Индуцированная подгонка может повысить точность молекулярного распознавания в присутствии конкуренции и шума через конформационная корректура механизм.[42]
Катализ
Ферменты могут ускорять реакции несколькими способами, каждый из которых снижает энергия активации (ΔG‡, Свободная энергия Гиббса )[43]
- Путем стабилизации переходного состояния:
- Создание среды с распределением заряда, дополнительным к переходному состоянию, чтобы снизить его энергию[44]
- Предоставляя альтернативный путь реакции:
- Временное взаимодействие с подложкой с образованием ковалентного промежуточного соединения для обеспечения переходного состояния с более низкой энергией[45]
- Дестабилизируя основное состояние подложки:
- Искажение связанного субстрата (субстратов) в их форму переходного состояния для уменьшения энергии, необходимой для достижения переходного состояния[46]
- Ориентируя подложки в продуктивном порядке, чтобы уменьшить реакцию энтропия изменять[47] (вклад этого механизма в катализ относительно невелик)[48]
Ферменты могут использовать несколько из этих механизмов одновременно. Например, протеазы Такие как трипсин выполнить ковалентный катализ с помощью каталитическая триада, стабилизировать накопление заряда в переходных состояниях с помощью оксианионная дыра, полный гидролиз с использованием ориентированного водного субстрата.[49]
Динамика
Ферменты - это не жесткие статичные структуры; вместо этого они имеют сложные внутренние динамические движения, то есть движения частей структуры фермента, таких как отдельные аминокислотные остатки, группы остатков, образующие белковая петля или единица вторичная структура, или даже целую белковый домен. Эти движения вызывают конформационный ансамбль немного разных структур, которые пересекаются друг с другом в равновесие. Различные состояния в этом ансамбле могут быть связаны с разными аспектами функции фермента. Например, разные конформации фермента дигидрофолатредуктаза связаны со стадиями связывания субстрата, катализа, высвобождения кофактора и высвобождения продукта каталитического цикла,[50] в соответствии с теория каталитического резонанса.
Презентация субстрата
Презентация субстрата это процесс, при котором фермент изолируется от его субстрата. Ферменты могут быть изолированы на плазматической мембране от субстрата в ядре или цитозоле. Или внутри мембраны фермент может быть изолирован в липидных плотах от своего субстрата в неупорядоченной области. Когда фермент высвобождается, он смешивается со своим субстратом. В качестве альтернативы фермент может быть изолирован рядом с его субстратом для активации фермента. Например, фермент может быть растворимым и после активации связываться с липидом в плазматической мембране, а затем действовать на молекулы в плазматической мембране.
Аллостерическая модуляция
Аллостерические сайты - это карманы на ферменте, отличные от активного сайта, которые связываются с молекулами в клеточной среде. Затем эти молекулы вызывают изменение конформации или динамики фермента, который преобразуется в активный центр, и таким образом влияет на скорость реакции фермента.[51] Таким образом, аллостерические взаимодействия могут либо ингибировать, либо активировать ферменты. Аллостерические взаимодействия с метаболитами выше или ниже по течению метаболического пути фермента вызывают Обратная связь регуляция, изменяя активность фермента в соответствии с поток через остальную часть пути.[52]
Кофакторы

Некоторым ферментам не требуются дополнительные компоненты для полной активности. Другие требуют, чтобы небелковые молекулы, называемые кофакторами, были связаны для активности.[53] Кофакторы могут быть либо неорганический (например., ионы металлов и железо-серные кластеры ) или же органические соединения (например., флавин и гем ). Эти кофакторы служат многим целям; например, ионы металлов могут помочь в стабилизации нуклеофильных частиц в активном центре.[54] Органические кофакторы могут быть либо коферменты, которые высвобождаются из активного центра фермента во время реакции, или протезные группы, которые прочно связаны с ферментом. Органические простетические группы могут быть связаны ковалентно (например, биотин в ферментах, таких как пируваткарбоксилаза ).[55]
Примером фермента, содержащего кофактор, является карбоангидраза, который использует кофактор цинка, связанный как часть его активного сайта.[56] Эти прочно связанные ионы или молекулы обычно находятся в активном центре и участвуют в катализе.[1]:8.1.1 Например, кофакторы флавина и гема часто участвуют в редокс реакции.[1]:17
Ферменты, требующие кофактора, но не имеющие одной связи, называются апоферменты или же апопротеины. Фермент вместе с кофактором (ами), необходимым для активности, называется холоэнзим (или галофермент). Период, термин холоэнзим также может применяться к ферментам, которые содержат несколько белковых субъединиц, таких как ДНК-полимеразы; здесь холофермент - это полный комплекс, содержащий все субъединицы, необходимые для активности.[1]:8.1.1
Коферменты
Коферменты - это небольшие органические молекулы, которые могут быть слабо или прочно связаны с ферментом. Коферменты переносят химические группы от одного фермента к другому.[57] Примеры включают НАДН, НАДФН и аденозинтрифосфат (АТФ). Некоторые коферменты, такие как флавинмононуклеотид (FMN), флавинаденин динуклеотид (FAD), пирофосфат тиамина (TPP) и тетрагидрофолат (THF), получены из витамины. Эти коферменты не могут быть синтезированы организмом de novo и близкородственные соединения (витамины) должны поступать с пищей. Переносимые химические группы включают:
- то гидрид ион (H−), принесенный НАД или НАДФ+
- фосфатная группа, переносимая аденозинтрифосфат
- ацетильная группа, переносимая кофермент А
- формильные, метенильные или метильные группы, переносимые фолиевая кислота и
- метильная группа, переносимая S-аденозилметионин[57]
Так как коферменты химически изменяются в результате действия ферментов, полезно рассматривать коферменты как особый класс субстратов или вторые субстраты, которые являются общими для многих различных ферментов. Например, известно около 1000 ферментов, использующих кофермент НАДН.[58]
Коферменты обычно непрерывно регенерируются, и их концентрация внутри клетки поддерживается на постоянном уровне. Например, НАДФН регенерируется через пентозофосфатный путь и S-аденозилметионин метионин аденозилтрансфераза. Эта непрерывная регенерация означает, что небольшие количества коферментов могут использоваться очень интенсивно. Например, человеческое тело ежедневно перерабатывает собственный вес АТФ.[59]
Термодинамика
Как и все катализаторы, ферменты не изменяют положение химического равновесия реакции. В присутствии фермента реакция протекает в том же направлении, что и без фермента, только быстрее.[1]:8.2.3 Например, карбоангидраза катализирует свою реакцию в любом направлении в зависимости от концентрации реагентов:[60]
- (в ткани; высокий CO2 концентрация)
(1)
![{displaystyle {ce {CO2 {} + H2O -> [{ext {Карбоангидраза}}] H2CO3}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/cb4c8837b26e96fe552c17d863f93e0618cd998b)
- (в легкие; низкий CO2 концентрация)
(2)
![{displaystyle {ce {CO2 {} + H2O <- [{ext {Карбоангидраза}}] H2CO3}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/618e95485aa1c3c44a29c557ac448ae5b544ff07)
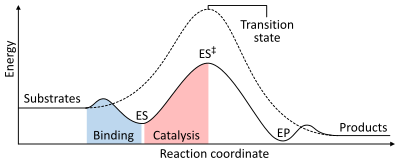
Скорость реакции зависит от энергия активации необходимо для формирования переходное состояние который затем распадается на продукты. Ферменты увеличивают скорость реакции за счет снижения энергии переходного состояния. Во-первых, связывание образует низкоэнергетический комплекс фермент-субстрат (ES). Во-вторых, фермент стабилизирует переходное состояние, так что для его достижения требуется меньше энергии по сравнению с некаталитической реакцией (ES‡). Наконец, комплекс фермент-продукт (EP) диссоциирует с высвобождением продуктов.[1]:8.3
Ферменты могут соединять две или более реакций, так что термодинамически благоприятная реакция может использоваться для «управления» термодинамически неблагоприятной, так что объединенная энергия продуктов ниже, чем у субстратов. Например, гидролиз АТФ часто используется для управления другими химическими реакциями.[61]
Кинетика


Кинетика ферментов - это исследование того, как ферменты связывают субстраты и превращают их в продукты.[62] Данные скорости, используемые в кинетическом анализе, обычно получают из ферментные анализы. В 1913 г. Леонор Михаэлис и Мод Леонора Ментен предложил количественную теорию кинетики ферментов, получившую название Кинетика Михаэлиса – Ментен.[63] Главный вклад Михаэлиса и Ментен заключался в том, что ферментативные реакции разделялись на две стадии. В первом субстрат обратимо связывается с ферментом, образуя комплекс фермент-субстрат. В их честь это иногда называют комплексом Михаэлиса-Ментен. Затем фермент катализирует химическую стадию реакции и высвобождает продукт. Эта работа получила дальнейшее развитие Г. Э. Бриггс и Дж. Б. С. Холдейн, который вывел кинетические уравнения, которые широко используются до сих пор.[64]
Нормы ферментов зависят от решение условия и субстрат концентрация. Чтобы определить максимальную скорость ферментативной реакции, концентрацию субстрата увеличивают до тех пор, пока не будет наблюдаться постоянная скорость образования продукта. Это показано на кривой насыщенности справа. Насыщение происходит потому, что по мере увеличения концентрации субстрата все больше и больше свободного фермента превращается в связанный с субстратом комплекс ES. При максимальной скорости реакции (VМаксимум) фермента все активные центры фермента связаны с субстратом, а количество комплекса ES является таким же, как общее количество фермента.[1]:8.4
VМаксимум является лишь одним из нескольких важных кинетических параметров. Также важно количество субстрата, необходимое для достижения заданной скорости реакции. Это дано Константа Михаэлиса-Ментен (Kм), которая представляет собой концентрацию субстрата, необходимую для того, чтобы фермент достиг половины максимальной скорости реакции; как правило, каждый фермент имеет характеристику KM для данного субстрата. Еще одна полезная константа: kКот, также называемый номер оборота, который представляет собой количество молекул субстрата, обрабатываемых одним активным центром в секунду.[1]:8.4
Эффективность фермента можно выразить через kКот/Kм. Это также называется константой специфичности и включает константы скорости для всех этапов реакции вплоть до первого необратимого шага. Поскольку константа специфичности отражает как сродство, так и каталитическую способность, она полезна для сравнения различных ферментов друг с другом или одного и того же фермента с разными субстратами. Теоретический максимум константы специфичности называется диффузионным пределом и составляет около 108 до 109 (M−1 s−1). В этот момент каждое столкновение фермента с его субстратом приводит к катализу, и скорость образования продукта ограничивается не скоростью реакции, а скоростью диффузии. Ферменты с этим свойством называются каталитически совершенный или же кинетически совершенный. Примером таких ферментов являются триозофосфат изомераза, карбоангидраза, ацетилхолинэстераза, каталаза, фумараза, β-лактамаза, и супероксиддисмутаза.[1]:8.4.2 Оборот таких ферментов может достигать нескольких миллионов реакций в секунду.[1]:9.2 Но большинство ферментов далеки от совершенства: средние значения и о и , соответственно.[65]




Кинетика Михаэлиса – Ментен опирается на закон массового действия, который выводится из предположений о свободном распространение и термодинамически обусловленное случайное столкновение. Многие биохимические или клеточные процессы значительно отклоняются от этих условий из-за макромолекулярное скопление и ограниченное движение молекул.[66] Более поздние сложные расширения модели пытаются исправить эти эффекты.[67]
Торможение

Скорость ферментативной реакции можно снизить различными типами ингибиторы ферментов.[69]:73–74
Типы торможения
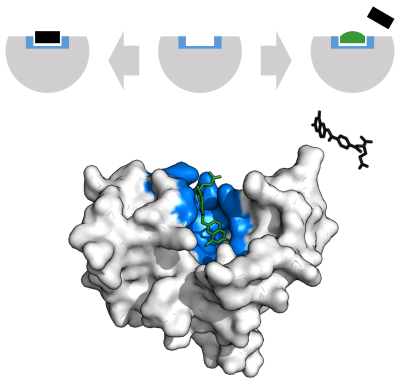
Конкурентный
А конкурентный ингибитор и субстрат не может одновременно связываться с ферментом.[70] Часто конкурентные ингибиторы сильно напоминают реальный субстрат фермента. Например, препарат метотрексат конкурентный ингибитор фермента дигидрофолатредуктаза, что катализирует снижение дигидрофолат к тетрагидрофолату.[68] Сходство структур дигидрофолата и этого препарата показано на прилагаемом рисунке. Этот тип ингибирования можно преодолеть с помощью высокой концентрации субстрата. В некоторых случаях ингибитор может связываться с сайтом, отличным от сайта связывания обычного субстрата, и оказывать аллостерический эффект изменить форму обычного места привязки.[71]
Неконкурентоспособный
А неконкурентный ингибитор связывается с сайтом, отличным от того, где связывается субстрат. Субстрат по-прежнему связывается со своим обычным сродством и, следовательно, Kм остается такой же. Однако ингибитор снижает каталитическую эффективность фермента, так что VМаксимум уменьшен. В отличие от конкурентного ингибирования неконкурентное ингибирование не может быть преодолено высокой концентрацией субстрата.[69]:76–78
Неконкурентоспособен
An неконкурентоспособный ингибитор не может связываться со свободным ферментом, только с комплексом фермент-субстрат; следовательно, эти типы ингибиторов наиболее эффективны при высокой концентрации субстрата. В присутствии ингибитора комплекс фермент-субстрат неактивен.[69]:78 Этот тип торможения встречается редко.[72]
Смешанный
А смешанный ингибитор связывается с аллостерическим сайтом, и связывание субстрата и ингибитора влияет друг на друга. Функция фермента снижается, но не устраняется при связывании с ингибитором. Этот тип ингибитора не подчиняется уравнению Михаэлиса-Ментен.[69]:76–78
Необратимый
An необратимый ингибитор навсегда инактивирует фермент, обычно за счет образования Ковалентная связь к белку.[73] Пенициллин[74] и аспирин[75] распространенные препараты, которые действуют подобным образом.
Функции ингибиторов
У многих организмов ингибиторы могут действовать как часть Обратная связь механизм. Если фермент производит слишком много одного вещества в организме, это вещество может действовать как ингибитор фермента в начале пути, который его производит, заставляя производство вещества замедляться или прекращаться, когда его достаточно. Это форма негативный отзыв. Основные метаболические пути, такие как цикл лимонной кислоты используйте этот механизм.[1]:17.2.2
Поскольку ингибиторы модулируют функцию ферментов, их часто используют в качестве лекарств. Многие такие препараты являются обратимыми конкурентными ингибиторами, которые напоминают нативный субстрат фермента, как и метотрексат над; другие известные примеры включают статины используется для лечения высоких холестерин,[76] и ингибиторы протеазы используется для лечения ретровирусный инфекции, такие как ВИЧ.[77] Типичный пример необратимого ингибитора, который используется в качестве лекарства: аспирин, что подавляет СОХ-1 и СОХ-2 ферменты, которые производят воспаление посыльный простагландин.[75] Другие ингибиторы ферментов - яды. Например, яд цианид необратимый ингибитор фермента, который соединяется с медью и железом в активном центре фермента цитохром с оксидаза и блоки клеточное дыхание.[78]
Факторы, влияющие на активность ферментов
Поскольку ферменты состоят из белков, их действие чувствительно к изменению многих физико-химических факторов, таких как pH, температура, концентрация субстрата и т. Д.
В следующей таблице показаны оптимальные значения pH для различных ферментов.[79]
| Фермент | Оптимальный pH | Описание pH |
|---|---|---|
| Пепсин | 1.5–1.6 | Очень кислая |
| Инвертаза | 4.5 | Кислая |
| Липаза (желудок) | 4.0–5.0 | Кислая |
| Липаза (касторовое масло) | 4.7 | Кислая |
| Липаза (поджелудочная железа) | 8.0 | Щелочной |
| Амилаза (солод) | 4.6–5.2 | Кислая |
| Амилаза (поджелудочная железа) | 6.7–7.0 | Кислотно-нейтральный |
| Целлобиаза | 5.0 | Кислая |
| Мальтаза | 6.1–6.8 | Кислая |
| Sucrase | 6.2 | Кислая |
| Каталаза | 7.0 | Нейтральный |
| Уреаза | 7.0 | Нейтральный |
| Холинэстераза | 7.0 | Нейтральный |
| Рибонуклеаза | 7.0–7.5 | Нейтральный |
| Фумараза | 7.8 | Щелочной |
| Трипсин | 7.8–8.7 | Щелочной |
| Аденозинтрифосфат | 9.0 | Щелочной |
| Аргиназа | 10.0 | Сильнощелочная |
Биологическая функция
Ферменты служат широкому спектру функции внутри живых организмов. Они незаменимы для преобразование сигнала и клеточная регуляция, часто через киназы и фосфатазы.[80] Они также генерируют движение, с миозин гидролизовать АТФ с образованием сокращение мышц, а также транспортировать грузы по ячейке в составе цитоскелет.[81] Другие АТФазы в клеточной мембране: ионные насосы участвует в активный транспорт. Ферменты также участвуют в более экзотических функциях, таких как люцифераза свет в светлячки.[82] Вирусы может также содержать ферменты для заражения клеток, такие как Интеграза ВИЧ и обратная транскриптаза, или для высвобождения вируса из клеток, например грипп вирус нейраминидаза.[83]
Важная функция ферментов заключается в пищеварительная система животных. Ферменты, такие как амилазы и протеазы разрушать большие молекулы (крахмал или же белки соответственно) на более мелкие, чтобы они могли всасываться в кишечнике. Молекулы крахмала, например, слишком велики для всасывания из кишечника, но ферменты гидролизуют цепи крахмала на более мелкие молекулы, такие как мальтоза и в конце концов глюкоза, который затем может абсорбироваться. Разные ферменты переваривают разные пищевые вещества. В жвачные животные, который имеет травоядный диеты, микроорганизмы в кишечнике производят другой фермент, целлюлаза, чтобы разрушить клеточные стенки целлюлозы растительного волокна.[84]
Метаболизм

Несколько ферментов могут работать вместе в определенном порядке, создавая метаболические пути.[1]:30.1 В метаболическом пути один фермент принимает продукт другого фермента в качестве субстрата. После каталитической реакции продукт передается другому ферменту. Иногда более одного фермента могут катализировать одну и ту же реакцию параллельно; это может позволить более сложное регулирование: например, с низкой постоянной активностью, обеспечиваемой одним ферментом, но индуцибельной высокой активностью со стороны второго фермента.[85]
Ферменты определяют, какие шаги происходят в этих путях. Без ферментов метаболизм не продвигался бы по одним и тем же этапам и не мог бы регулироваться для удовлетворения потребностей клетки. Большинство центральных метаболических путей регулируются на нескольких ключевых этапах, обычно с помощью ферментов, активность которых включает гидролиз АТФ. Поскольку эта реакция высвобождает так много энергии, другие реакции термодинамически неблагоприятный может быть связан с гидролизом АТФ, управляя общей серией связанных метаболических реакций.[1]:30.1
Контроль активности
Существует пять основных способов контроля активности ферментов в клетке.[1]:30.1.1
Регулирование
Ферменты могут быть либо активирован или же подавленный другими молекулами. Например, конечный продукт (продукты) метаболического пути часто является ингибитором одного из первых ферментов этого пути (обычно первая необратимая стадия, называемая коммитированной стадией), таким образом регулируя количество конечного продукта, производимого этими путями. Такой регуляторный механизм называется механизм отрицательной обратной связи, потому что количество производимого конечного продукта регулируется его собственной концентрацией.[86]:141–48 Механизм отрицательной обратной связи может эффективно регулировать скорость синтеза промежуточных метаболитов в соответствии с потребностями клеток. Это способствует эффективному распределению материалов и экономии энергии, а также предотвращает избыточное производство конечной продукции. Как и другие гомеостатические устройства, контроль ферментативного действия помогает поддерживать стабильную внутреннюю среду в живых организмах.[86]:141
Посттрансляционная модификация
Примеры посттрансляционная модификация включают фосфорилирование, миристоилирование и гликозилирование.[86]:149–69 Например, в ответ на инсулин, то фосфорилирование множества ферментов, включая гликогенсинтаза, помогает контролировать синтез или деградацию гликоген и позволяет клетке реагировать на изменения в содержание сахара в крови.[87] Другой пример посттрансляционной модификации - это расщепление полипептидной цепи. Химотрипсин, пищеварительный протеаза, производится в неактивной форме как химотрипсиноген в поджелудочная железа и транспортируется в таком виде на желудок где он активируется. Это мешает ферменту переваривать поджелудочную железу или другие ткани до того, как он попадет в кишечник. Этот тип неактивного предшественника фермента известен как зимоген[86]:149–53 или профермент.
Количество
Производство ферментов (транскрипция и перевод ферментных генов) может усиливаться или уменьшаться клеткой в ответ на изменения в окружающей среде клетки. Эта форма генная регуляция называется индукция ферментов. Например, бактерии могут стать устойчив к антибиотикам Такие как пенициллин потому что ферменты называются бета-лактамазы индуцируются, что гидролизуют важнейшие бета-лактамное кольцо внутри молекулы пенициллина.[88] Другой пример - ферменты в печень называется оксидазы цитохрома P450, которые важны в метаболизм лекарств. Индукция или ингибирование этих ферментов может вызвать лекарственные взаимодействия.[89] Уровни ферментов также можно регулировать, изменяя скорость ферментов. деградация.[1]:30.1.1 Противоположностью индукции фермента является ферментная репрессия.
Субклеточное распределение
Ферменты могут быть разделены на части, при этом разные метаболические пути происходят в разных клеточные отсеки. Например, жирные кислоты синтезируются одним набором ферментов в цитозоль, эндоплазматический ретикулум и Гольджи и используется другим набором ферментов в качестве источника энергии в митохондрия, через β-окисление.[90] Кроме того, торговля людьми фермента в разные компартменты может изменить степень протонирование (например, нейтральный цитоплазма и кислый лизосома ) или в окислительном состоянии (например, окислительный периплазма или сокращение цитоплазма ), что, в свою очередь, влияет на активность ферментов.[91] В отличие от разделения на мембраносвязанные органеллы, субклеточная локализация фермента также может быть изменена путем полимеризации ферментов в макромолекулярные цитоплазматические филаменты.[92][93]
Специализация организации
В многоклеточный эукариоты, ячейки в разных органы и ткани иметь разные модели экспрессия гена и поэтому имеют разные наборы ферментов (известных как изоферменты ) доступны для метаболических реакций. Это обеспечивает механизм регулирования общего метаболизма организма. Например, гексокиназа, первый фермент в гликолиз путь, имеет специальную форму, называемую глюкокиназа выраженный в печень и поджелудочная железа что имеет более низкий близость для глюкозы еще более чувствителен к концентрации глюкозы.[94] Этот фермент участвует в восприятии содержание сахара в крови и регулирование инсулин производство.[95]
Участие в болезни

Поскольку строгий контроль активности ферментов важен для гомеостаз, любая неисправность (мутация, перепроизводство, недопроизводство или удаление) одного критического фермента может привести к генетическое заболевание. Неисправность только одного типа фермента из тысяч типов, присутствующих в организме человека, может быть фатальной. Пример фатального генетическое заболевание из-за недостаточности ферментов Болезнь Тея – Сакса, у которых у пациентов отсутствует фермент гексозаминидаза.[96][97]
Одним из примеров недостаточности ферментов является наиболее распространенный тип фенилкетонурия. Множество различных мутаций одной аминокислоты в ферменте фенилаланингидроксилаза, который катализирует первый шаг в деградации фенилаланин, приводят к накоплению фенилаланина и связанных с ним продуктов. Некоторые мутации находятся в активном центре, что напрямую нарушает связывание и катализ, но многие из них находятся далеко от активного центра и снижают активность, дестабилизируя структуру белка или влияя на правильную олигомеризацию.[98][99] Это может привести к Интеллектуальная недееспособность если болезнь не лечить.[100] Другой пример дефицит псевдохолинэстеразы, при которых нарушена способность организма расщеплять препараты на основе эфира холина.[101] Пероральное введение ферментов можно использовать для лечения некоторых функциональных дефицитов ферментов, таких как панкреатическая недостаточность[102] и непереносимость лактозы.[103]
Другой способ, которым сбой ферментов может вызвать заболевание, связан с мутации зародышевой линии в генах, кодирующих Ремонт ДНК ферменты. Дефекты этих ферментов вызывают рак, потому что клетки менее способны восстанавливать мутации в своих геномы. Это вызывает медленное накопление мутаций и приводит к развитие рака. Пример такой наследственной онкологический синдром является пигментная ксеродермия, что вызывает развитие рак кожи в ответ даже на минимальное воздействие ультрафиолетовый свет.[104][105]
Эволюция
Как и любой другой белок, ферменты со временем меняются мутации и расхождение последовательности. Учитывая их центральную роль в метаболизм, эволюция ферментов играет решающую роль в приспособление. Таким образом, ключевой вопрос заключается в том, могут ли ферменты одновременно изменять свою ферментативную активность и каким образом. Принято считать, что многие новые ферментативные активности развились благодаря дупликация гена и мутации повторяющихся копий, хотя эволюция также может происходить без дублирования. Одним из примеров фермента, изменившего свою активность, является предок метиониламинопептидаза (MAP) и креатинамидногидролаза (креатиназа ), которые явно гомологичны, но катализируют очень разные реакции (MAP удаляет аминоконцевой метионин в новых белках при гидролизе креатиназы креатин к саркозин и мочевина ). Кроме того, MAP зависит от ионов металлов, а креатиназа - нет, поэтому это свойство также было потеряно со временем.[106] Небольшие изменения ферментативной активности чрезвычайно распространены среди ферментов. В частности, специфичность связывания субстрата (см. Выше) может легко и быстро измениться при изменении одной аминокислоты в их карманах связывания субстрата. Это часто наблюдается в основных классах ферментов, таких как киназы.[107]
Искусственная (in vitro) эволюция в настоящее время широко используется для изменения активности или специфичности ферментов в промышленных целях (см. Ниже).
Промышленное применение
Ферменты используются в химическая индустрия и другие промышленные применения, когда требуются исключительно специфические катализаторы. Ферменты в целом ограничены по количеству реакций, которые они должны катализировать, а также из-за отсутствия стабильности в органические растворители и при высоких температурах. Как следствие, белковая инженерия является активной областью исследований и включает попытки создания новых ферментов с новыми свойствами либо посредством рационального дизайна, либо in vitro эволюция.[108][109] Эти усилия начали приносить успех, и теперь несколько ферментов были разработаны «с нуля», чтобы катализировать реакции, которые не происходят в природе.[110]
Смотрите также
Ферментные базы данных
Рекомендации
- ^ а б c d е ж грамм час я j k л м п о п q р s т ты Страйер Л., Берг Дж. М., Тимочко Дж. Л. (2002). Биохимия (5-е изд.). Сан-Франциско: W.H. Фримен. ISBN 0-7167-4955-6.

- ^ Мерфи Дж. М., Фархан Х., Айерс ПА (2017). «Био-зомби: рост псевдоферментов в биологии». Biochem Soc Trans. 45 (2): 537–544. Дои:10.1042 / bst20160400. PMID 28408493.
- ^ Мерфи Дж. М. и др. (2014). «Надежная методология подкласса псевдокиназ на основе их свойств связывания нуклеотидов». Биохимический журнал. 457 (2): 323–334. Дои:10.1042 / BJ20131174. ЧВК 5679212. PMID 24107129.
- ^ Schomburg I, Chang A, Placzek S, Söhngen C, Rother M, Lang M, Munaretto C, Ulas S, Stelzer M, Grote A, Scheer M, Schomburg D (январь 2013 г.). «BRENDA в 2013 году: интегрированные реакции, кинетические данные, данные о функции ферментов, улучшенная классификация болезней: новые возможности и содержание в BRENDA». Исследования нуклеиновых кислот. 41 (Выпуск базы данных): D764–72. Дои:10.1093 / нар / gks1049. ЧВК 3531171. PMID 23203881.
- ^ Радзичка А., Вольфенден Р. (январь 1995 г.). «Опытный фермент». Наука. 267 (5194): 90–931. Bibcode:1995Научный ... 267 ... 90R. Дои:10.1126 / science.7809611. PMID 7809611. S2CID 8145198.
- ^ Каллахан Б.П., Миллер Б.Г. (декабрь 2007 г.). «Декарбоксилаза OMP - загадка сохраняется». Биоорганическая химия. 35 (6): 465–9. Дои:10.1016 / j.bioorg.2007.07.004. PMID 17889251.
- ^ де Реомюр РА (1752 г.). «Наблюдения за пищеварением уазов». Histoire de l'Académie Royale des Sciences. 1752: 266, 461.
- ^ Уильямс HS (1904). История науки: в пяти томах. Том IV: Современное развитие химических и биологических наук. Харпер и братья.
- ^ Payen A, Persoz JF (1833). "Памятка о диастазе, основных продуктах взаимодействия и различных приложениях для промышленности" [Воспоминания о диастазе, основных продуктах ее реакций и их применении в промышленном искусстве]. Анналы химии и тела. 2-й (на французском). 53: 73–92.
- ^ Манчестер KL (декабрь 1995 г.). «Луи Пастер (1822–1895) - случайность и подготовленность». Тенденции в биотехнологии. 13 (12): 511–5. Дои:10.1016 / S0167-7799 (00) 89014-9. PMID 8595136.
- ^ Кюне ввел слово «фермент» в: Кюне W (1877 г.). "Über das Verhalten verschiedener organisirter und sog. Ungeformter Fermente" [О поведении различных организованных и так называемых несформированных ферментов]. Verhandlungen des Naturhistorisch-medicinischen Vereins zu Heidelberg. новая серия (на немецком языке). 1 (3): 190–193. Соответствующий отрывок на странице 190: "Um Missverständnissen vorzubeugen und lästige Umschreibungen zu vermeiden schlägt Vortragender vor, die ungeformten oder nicht organisirten Fermente, deren Wirkung ohne Anwesenheit von Organismen und ausserhalb derseln erfolgen Фермент zu bezeichnen ". (Перевод: Чтобы избежать недоразумений и избежать громоздких перифраз, [автор, преподаватель университета] предлагает обозначать как «ферменты» несформированные или неорганизованные ферменты, действие которых может происходить без присутствия организмов и вне их.)
- ^ Холмс FL (2003). «Ферменты». В Heilbron JL (ред.). Оксфордский компаньон по истории современной науки. Оксфорд: Издательство Оксфордского университета. п. 270. ISBN 9780199743766.
- ^ "Эдуард Бухнер". Биография Нобелевского лауреата. Nobelprize.org. Получено 23 февраля 2015.
- ^ «Эдуард Бюхнер - Нобелевская лекция: Бесклеточная ферментация». Nobelprize.org. 1907. Получено 23 февраля 2015.
- ^ Название ферментов путем добавления суффикса «-аза» к субстрату, на котором действует фермент, было возведено французским ученым. Эмиль Дюкло (1840–1904), которые намеревались почтить память первооткрывателей диастаза - первый выделенный фермент - представив эту практику в своей книге Duclaux E (1899). Traité de microbiologie: диастазы, токсины и вены [Трактат по микробиологии: диастазы, токсины и яды] (На французском). Париж, Франция: Masson and Co. См. Главу 1, особенно страницу 9.
- ^ Willstätter R (1927). «Лекция Фарадея. Проблемы и методы исследования ферментов». Журнал химического общества (возобновлено): 1359–1381. Дои:10.1039 / JR9270001359. цитируется в Удар D (апрель 2000 г.). «Итак, мы понимаем, как работают ферменты?» (PDF). Структура. 8 (4): R77 – R81. Дои:10.1016 / S0969-2126 (00) 00125-8. PMID 10801479. Архивировано из оригинал (PDF) 4 марта 2016 г.. Получено 16 февраля 2012.
- ^ "Нобелевские премии и лауреаты: Нобелевская премия по химии 1946 г.". Nobelprize.org. Получено 23 февраля 2015.
- ^ Блейк С.К., Кениг Д.Ф., Майр Г.А., Северный АС, Филлипс, округ Колумбия, Сарма В.Р. (май 1965 г.). «Структура лизоцима куриного яйца-белка. Трехмерный синтез Фурье с разрешением 2 Ангстрема». Природа. 206 (4986): 757–61. Bibcode:1965Натура.206..757Б. Дои:10.1038 / 206757a0. PMID 5891407. S2CID 4161467.
- ^ Джонсон Л.Н., Петско Г.А. (1999). «Дэвид Филлипс и происхождение структурной энзимологии». Trends Biochem. Наука. 24 (7): 287–9. Дои:10.1016 / S0968-0004 (99) 01423-1. PMID 10390620.
- ^ Номенклатурный комитет. «Классификация и номенклатура ферментов по реакциям, которые они катализируют». Международный союз биохимии и молекулярной биологии (NC-IUBMB). Школа биологических и химических наук Королевы Марии Лондонского университета. Архивировано из оригинал 17 марта 2015 г.. Получено 6 марта 2015.
- ^ Номенклатурный комитет. «EC 2.7.1.1». Международный союз биохимии и молекулярной биологии (NC-IUBMB). Школа биологических и химических наук Королевы Марии Лондонского университета. Архивировано из оригинал 1 декабря 2014 г.. Получено 6 марта 2015.
- ^ Анфинсен CB (июль 1973 г.). «Принципы, регулирующие складывание белковых цепей». Наука. 181 (4096): 223–30. Bibcode:1973Sci ... 181..223A. Дои:10.1126 / science.181.4096.223. PMID 4124164.
- ^ Данауэй-Мариано Д. (ноябрь 2008 г.). «Открытие ферментной функции». Структура. 16 (11): 1599–600. Дои:10.1016 / j.str.2008.10.001. PMID 19000810.
- ^ Пецко Г.А., Ринге Д. (2003). «Глава 1: От последовательности к структуре». Структура и функция белка. Лондон: Новая наука. п. 27. ISBN 978-1405119221.
- ^ Chen LH, Kenyon GL, Curtin F, Harayama S, Bembenek ME, Hajipour G, Whitman CP (сентябрь 1992 г.). «4-оксалокротонаттаутомераза, фермент, состоящий из 62 аминокислотных остатков на мономер». Журнал биологической химии. 267 (25): 17716–21. PMID 1339435.
- ^ Смит С. (декабрь 1994 г.). «Синтаза жирных кислот животных: один ген, один полипептид, семь ферментов». Журнал FASEB. 8 (15): 1248–59. Дои:10.1096 / fasebj.8.15.8001737. PMID 8001737. S2CID 22853095.
- ^ "Атлас каталитических мест". Европейский институт биоинформатики. Получено 4 апреля 2007.
- ^ а б Suzuki H (2015). «Глава 7: Активная структура сайта». Как работают ферменты: от структуры к функции. Бока-Ратон, Флорида: CRC Press. С. 117–140. ISBN 978-981-4463-92-8.
- ^ Краусс Г (2003). «Регламент активности ферментов». Биохимия передачи и регуляции сигналов (3-е изд.). Вайнхайм: Wiley-VCH. С. 89–114. ISBN 9783527605767.
- ^ Jaeger KE, Eggert T (август 2004 г.). «Энантиоселективный биокатализ, оптимизированный направленной эволюцией». Текущее мнение в области биотехнологии. 15 (4): 305–13. Дои:10.1016 / j.copbio.2004.06.007. PMID 15358000.
- ^ Шевелев И.В., Хюбшер У. (май 2002 г.). «Экзонуклеазы 3 '5'». Обзоры природы Молекулярная клеточная биология. 3 (5): 364–76. Дои:10.1038 / nrm804. PMID 11988770. S2CID 31605786.
- ^ Зенкин Н., Юзенкова Ю., Северинов К. (июль 2006 г.). «Корректура транскрипции с помощью транскрипции». Наука. 313 (5786): 518–20. Bibcode:2006Научный ... 313..518Z. Дои:10.1126 / science.1127422. PMID 16873663. S2CID 40772789.
- ^ Ибба М., Солл Д. (2000). «Синтез аминоацил-тРНК». Ежегодный обзор биохимии. 69: 617–50. Дои:10.1146 / annurev.biochem.69.1.617. PMID 10966471.
- ^ Роднина М.В., Винтермейер В. (2001). «Верность отбора аминоацил-тРНК на рибосоме: кинетические и структурные механизмы». Ежегодный обзор биохимии. 70: 415–35. Дои:10.1146 / annurev.biochem.70.1.415. PMID 11395413.
- ^ Херсонский О, Тауфик Д.С. (2010). «Ферментная неразборчивость: механистическая и эволюционная перспектива». Ежегодный обзор биохимии. 79: 471–505. Дои:10.1146 / annurev-biochem-030409-143718. PMID 20235827.
- ^ О'Брайен П.Дж., Хершлаг Д. (апрель 1999 г.). «Каталитическая неразборчивость и эволюция новых ферментативных активностей». Химия и биология. 6 (4): R91 – R105. Дои:10.1016 / S1074-5521 (99) 80033-7. PMID 10099128.
- ^ Фишер Э (1894). "Einfluss der Configuration auf die Wirkung der Enzyme" [Влияние конфигурации на действие ферментов]. Berichte der Deutschen Chemischen Gesellschaft zu Berlin (на немецком). 27 (3): 2985–93. Дои:10.1002 / cber.18940270364. Со страницы 2992: "Um ein Bild zu gebrauchen, will ich sagen, dass Enzym und Glucosid wie Schloss und Schlüssel zu einander passen müssen, um eine chemische Wirkung auf einander ausüben zu können". (Чтобы использовать изображение, я скажу, что фермент и глюкозид [т.е. производное глюкозы] должны соответствовать друг другу, как замок и ключ, чтобы иметь возможность оказывать химическое воздействие друг на друга.)
- ^ Купер GM (2000). «Глава 2.2: Центральная роль ферментов как биологических катализаторов». Клетка: молекулярный подход (2-е изд.). Вашингтон (округ Колумбия): ASM Press. ISBN 0-87893-106-6.
- ^ Кошланд Д.Е. (февраль 1958 г.). «Применение теории ферментной специфичности к синтезу белков». Труды Национальной академии наук Соединенных Штатов Америки. 44 (2): 98–104. Bibcode:1958ПНАС ... 44 ... 98К. Дои:10.1073 / пнас.44.2.98. ЧВК 335371. PMID 16590179.
- ^ Васелла А., Дэвис Г. Дж., Бём М. (октябрь 2002 г.). «Гликозидазные механизмы». Современное мнение в области химической биологии. 6 (5): 619–29. Дои:10.1016 / S1367-5931 (02) 00380-0. PMID 12413546.
- ^ Бойер Р. (2002). «Глава 6: Ферменты I, реакции, кинетика и ингибирование». Понятия в биохимии (2-е изд.). Нью-Йорк, Чичестер, Вайнхайм, Брисбен, Сингапур, Торонто: John Wiley & Sons, Inc., стр. 137–8. ISBN 0-470-00379-0. OCLC 51720783.
- ^ Савир Ю., Тласти Т. (2007). Скалас Э (ред.). «Конформационная корректура: влияние конформационных изменений на специфичность молекулярного распознавания» (PDF). PLOS ONE. 2 (5): e468. Bibcode:2007PLoSO ... 2..468S. Дои:10.1371 / journal.pone.0000468. ЧВК 1868595. PMID 17520027. Архивировано из оригинал (PDF) 14 мая 2011 г.. Получено 22 августа 2010.
- ^ Фершт А (1985). Структура и механизм фермента. Сан-Франциско: W.H. Фримен. С. 50–2. ISBN 978-0-7167-1615-0.
- ^ Варшел А., Шарма П. К., Като М., Сян Ю., Лю Х., Олссон М. Х. (август 2006 г.). «Электростатическая основа ферментативного катализа». Химические обзоры. 106 (8): 3210–35. Дои:10.1021 / cr0503106. PMID 16895325.
- ^ Кокс ММ, Нельсон Д.Л. (2013). «Глава 6.2: Как работают ферменты». Принципы биохимии Ленингера (6-е изд.). Нью-Йорк, Нью-Йорк: W.H. Фримен. п. 195. ISBN 978-1464109621.
- ^ Бенкович SJ, Hammes-Schiffer S (август 2003 г.). «Перспектива ферментного катализа». Наука. 301 (5637): 1196–202. Bibcode:2003Научный ... 301.1196B. Дои:10.1126 / science.1085515. PMID 12947189. S2CID 7899320.
- ^ Дженкс WP (1987). Катализ в химии и энзимологии. Минеола, Нью-Йорк: Дувр. ISBN 978-0-486-65460-7.
- ^ Вилла Дж., Страйбл М., Гленнон Т.М., Шам Ю.Й., Чу З.Т., Варшел А. (октябрь 2000 г.). "Насколько важны энтропийные вклады в ферментативный катализ?". Труды Национальной академии наук Соединенных Штатов Америки. 97 (22): 11899–904. Bibcode:2000PNAS ... 9711899V. Дои:10.1073 / пнас.97.22.11899. ЧВК 17266. PMID 11050223.
- ^ Польгар, Л. (7 июля 2005 г.). «Каталитическая триада сериновых пептидаз». Клеточные и молекулярные науки о жизни. 62 (19–20): 2161–2172. Дои:10.1007 / s00018-005-5160-х. ISSN 1420-682X. PMID 16003488. S2CID 3343824.
- ^ Раманатан А., Савол А., Бургер В., Ченнубхотла К.С., Агарвал П.К. (2014). «Белковые конформационные популяции и функционально значимые подсостояния». Соотв. Chem. Res. 47 (1): 149–56. Дои:10.1021 / ar400084s. OSTI 1565147. PMID 23988159.
- ^ Цай С.Дж., Дель Соль А., Нусинов Р. (2009). «Белковая аллостерия, передача сигналов и динамика: классификационная схема аллостерических механизмов» (PDF). Мол Биосист. 5 (3): 207–16. Дои:10.1039 / b819720b. ЧВК 2898650. PMID 19225609.
- ^ Changeux JP, Edelstein SJ (июнь 2005 г.). «Аллостерические механизмы передачи сигналов». Наука. 308 (5727): 1424–8. Bibcode:2005Sci ... 308.1424C. Дои:10.1126 / science.1108595. PMID 15933191. S2CID 10621930.
- ^ де Болстер М. (1997). «Глоссарий терминов, используемых в биоинорганической химии: кофактор». Международный союз теоретической и прикладной химии. Архивировано из оригинал 21 января 2017 г.. Получено 30 октября 2007.
- ^ Воет Д., Воет Дж., Пратт С. (2016). Основы биохимии. Хобокен, Нью-Джерси: John Wiley & Sons, Inc., стр. 336. ISBN 978-1-118-91840-1.
- ^ Чепмен-Смит А., Кронан Дж. Э. (1999). «Ферментативное биотинилирование белков: посттрансляционная модификация исключительной специфичности». Trends Biochem. Наука. 24 (9): 359–63. Дои:10.1016 / s0968-0004 (99) 01438-3. PMID 10470036.
- ^ Фишер З., Эрнандес Прада Дж. А., Ту С., Дуда Д., Йошиока С., Ан Х, Говиндасами Л., Сильверман Д. Н., МакКенна Р. (февраль 2005 г.). «Структурная и кинетическая характеристика гистидина активного центра в качестве протонного челнока в катализе человеческой карбоангидразой II». Биохимия. 44 (4): 1097–115. Дои:10.1021 / bi0480279. PMID 15667203.
- ^ а б Вагнер А.Л. (1975). Витамины и коферменты. Krieger Pub Co. ISBN 0-88275-258-8.
- ^ "BRENDA - комплексная система информации о ферментах". Technische Universität Брауншвейг. Получено 23 февраля 2015.
- ^ Törnroth-Horsefield S, Neutze R (декабрь 2008 г.). «Открытие и закрытие ворот метаболита». Труды Национальной академии наук Соединенных Штатов Америки. 105 (50): 19565–6. Bibcode:2008ПНАС..10519565Т. Дои:10.1073 / pnas.0810654106. ЧВК 2604989. PMID 19073922.
- ^ МакАрдл В.Д., Катч Ф., Катч В.Л. (2006). «Глава 9: Легочная система и упражнения». Основы физиологии упражнений (3-е изд.). Балтимор, Мэриленд: Липпинкотт Уильямс и Уилкинс. С. 312–3. ISBN 978-0781749916.
- ^ Фергюсон С.Дж., Николлс Д., Фергюсон С. (2002). Биоэнергетика 3 (3-е изд.). Сан-Диего: академический. ISBN 0-12-518121-3.
- ^ Ганс, Биссвангер. Кинетика ферментов: принципы и методы (Третье, дополненное и дополненное ред.). Вайнхайм, Германия. ISBN 9783527806461. OCLC 992976641.
- ^ Михаэлис Л, Ментен М (1913). "Die Kinetik der Invertinwirkung" [Кинетика действия инвертазы]. Biochem. Z. (на немецком). 49: 333–369.; Михаэлис Л., Ментен М.Л., Джонсон К.А., Гуди Р.С. (2011). «Исходная константа Михаэлиса: перевод статьи Михаэлиса – Ментен 1913 года». Биохимия. 50 (39): 8264–9. Дои:10.1021 / bi201284u. ЧВК 3381512. PMID 21888353.
- ^ Бриггс Дж. Э., Холдейн Дж. Б. (1925). «Заметка о кинетике действия ферментов». Биохимический журнал. 19 (2): 338–9. Дои:10.1042 / bj0190338. ЧВК 1259181. PMID 16743508.
- ^ Бар-Эвен А., Нур Э, Савир Й, Либермейстер В., Давиди Д., Тауфик Д.С., Майло Р. (2011). «Умеренно эффективный фермент: эволюционные и физико-химические тенденции, определяющие параметры фермента». Биохимия. 50 (21): 4402–10. Дои:10.1021 / bi2002289. PMID 21506553.
- ^ Эллис Р.Дж. (октябрь 2001 г.). «Макромолекулярная скученность: очевидно, но недооценивается». Тенденции в биохимических науках. 26 (10): 597–604. Дои:10.1016 / S0968-0004 (01) 01938-7. PMID 11590012.
- ^ Копельман Р. (сентябрь 1988 г.). «Кинетика фрактальных реакций». Наука. 241 (4873): 1620–26. Bibcode:1988Научный ... 241.1620K. Дои:10.1126 / science.241.4873.1620. PMID 17820893. S2CID 23465446.
- ^ а б Гудселл, Дэвид С. (1 августа 1999 г.). «Молекулярная перспектива: метотрексат». Онколог. 4 (4): 340–341. Дои:10.1634 / теонколог.4-4-340. ISSN 1083-7159. PMID 10476546.
- ^ а б c d Корниш-Боуден А (2004). Основы кинетики ферментов (3-е изд.). Лондон: Портленд Пресс. ISBN 1-85578-158-1.
- ^ Прайс NC (1979). «Что подразумевается под« конкурентным торможением »?». Тенденции в биохимических науках. 4 (11): N272 – N273. Дои:10.1016/0968-0004(79)90205-6.
- ^ Ву П., Клаузен М.Х., Нильсен Т.Э. (декабрь 2015 г.). «Аллостерические низкомолекулярные ингибиторы киназы» (PDF). Фармакология и терапия. 156: 59–68. Дои:10.1016 / j.pharmthera.2015.10.002. PMID 26478442.
- ^ Корниш-Боуден А (Июль 1986 г.). «Почему неконкурентное торможение так редко? Возможное объяснение, имеющее значение для разработки лекарств и пестицидов». Письма FEBS. 203 (1): 3–6. Дои:10.1016/0014-5793(86)81424-7. PMID 3720956. S2CID 45356060.
- ^ Стрелов, Джон М. (1 января 2017 г.). «Перспектива кинетики ковалентного и необратимого ингибирования». SLAS DISCOVERY: Развитие исследований и разработок в области наук о жизни. 22 (1): 3–20. Дои:10.1177/1087057116671509. ISSN 2472-5552. PMID 27703080.
- ^ Фишер Дж. Ф., Меруэ С. О., Mobashery S (февраль 2005 г.). «Бактериальная резистентность к бета-лактамным антибиотикам: убедительный оппортунизм, неотразимая возможность». Химические обзоры. 105 (2): 395–424. Дои:10.1021 / cr030102i. PMID 15700950.
- ^ а б Джонсон Д.С., Вирапана Е., Краватт Б.Ф. (июнь 2010 г.). «Стратегии обнаружения и снижения рисков ковалентных необратимых ингибиторов ферментов». Медицинская химия будущего. 2 (6): 949–64. Дои:10.4155 / fmc.10.21. ЧВК 2904065. PMID 20640225.
- ^ Эндо А (1 ноября 1992 г.). «Открытие и разработка ингибиторов HMG-CoA редуктазы». J. Lipid Res. 33 (11): 1569–82. PMID 1464741.
- ^ Влодавер А., Вондрасек Дж. (1998). «Ингибиторы протеазы ВИЧ-1: главный успех разработки лекарств с использованием структуры». Ежегодный обзор биофизики и структуры биомолекул. 27: 249–84. Дои:10.1146 / annurev.biophys.27.1.249. PMID 9646869. S2CID 10205781.
- ^ Йошикава С., Каугей В.С. (май 1990 г.). «Инфракрасное свидетельство связывания цианида с участками железа и меди в цитохром с оксидазе сердца крупного рогатого скота. Значение восстановления кислорода». Журнал биологической химии. 265 (14): 7945–58. PMID 2159465.
- ^ Джайн, Дж. Л. (май 1999 г.). Основы биохимии. Нью-Дели: С. Чанд и Ко. ISBN 8121903432. OCLC 818809626.
- ^ Хантер Т. (январь 1995 г.). «Протеинкиназы и фосфатазы: инь и янь фосфорилирования белков и передачи сигналов». Клетка. 80 (2): 225–36. Дои:10.1016/0092-8674(95)90405-0. PMID 7834742. S2CID 13999125.
- ^ Берг Дж. С., Пауэлл BC, Чейни Р. Э. (апрель 2001 г.). «Тысячелетняя перепись миозина». Молекулярная биология клетки. 12 (4): 780–94. Дои:10.1091 / mbc.12.4.780. ЧВК 32266. PMID 11294886.
- ^ Мейген Э.А. (март 1991 г.). «Молекулярная биология бактериальной биолюминесценции». Микробиологические обзоры. 55 (1): 123–42. Дои:10.1128 / MMBR.55.1.123-142.1991. ЧВК 372803. PMID 2030669.
- ^ Де Клерк Э. (2002). «Основные моменты в разработке новых противовирусных агентов». Мини Rev Med Chem. 2 (2): 163–75. Дои:10.2174/1389557024605474. PMID 12370077.
- ^ Mackie RI, White BA (октябрь 1990 г.). «Последние достижения в области микробной экологии и метаболизма рубца: потенциальное влияние на выход питательных веществ». Журнал молочной науки. 73 (10): 2971–95. Дои:10.3168 / jds.S0022-0302 (90) 78986-2. PMID 2178174.
- ^ Rouzer CA, Марнетт LJ (2009). «Циклооксигеназы: структурное и функциональное понимание». J. Lipid Res. 50 Дополнение: S29–34. Дои:10.1194 / мл. R800042-JLR200. ЧВК 2674713. PMID 18952571.
- ^ а б c d Suzuki H (2015). «Глава 8: Контроль активности ферментов». Как работают ферменты: от структуры к функции. Бока-Ратон, Флорида: CRC Press. С. 141–69. ISBN 978-981-4463-92-8.
- ^ Doble BW, Woodgett JR (апрель 2003 г.). «ГСК-3: хитрости для многозадачной киназы». Журнал клеточной науки. 116 (Pt 7): 1175–86. Дои:10.1242 / jcs.00384. ЧВК 3006448. PMID 12615961.
- ^ Беннет П.М., Чопра I (1993). «Молекулярные основы индукции бета-лактамаз у бактерий». Антимикробный. Агенты Chemother. 37 (2): 153–8. Дои:10.1128 / aac.37.2.153. ЧВК 187630. PMID 8452343.
- ^ Скетт П., Гибсон Г.Г. (2001). «Глава 3: Индукция и ингибирование метаболизма лекарств». Введение в метаболизм лекарств (3-е изд.). Челтенхэм, Великобритания: Nelson Thornes Publishers. С. 87–118. ISBN 978-0748760114.
- ^ Фаэргеман Н. Дж., Кнудсен Дж. (Апрель 1997 г.). «Роль длинноцепочечных сложных эфиров ацил-КоА с длинной цепью в регуляции метаболизма и клеточной сигнализации». Биохимический журнал. 323 (Чт 1): 1–12. Дои:10.1042 / bj3230001. ЧВК 1218279. PMID 9173866.
- ^ Suzuki H (2015). «Глава 4: Влияние pH, температуры и высокого давления на ферментативную активность». Как работают ферменты: от структуры к функции. Бока-Ратон, Флорида: CRC Press. С. 53–74. ISBN 978-981-4463-92-8.
- ^ Нори С., Сато Б.К., Бройер Р.М., Вильгельм Дж. Э. (август 2010 г.). «Идентификация новых филамент-образующих белков у Saccharomyces cerevisiae и Drosophila melanogaster». Журнал клеточной биологии. 190 (4): 541–51. Дои:10.1083 / jcb.201003001. ЧВК 2928026. PMID 20713603.
- ^ Огхи Г.Н., Лю Дж.Л. (2015). «Метаболическая регуляция посредством ферментативной филаментации». Критические обзоры в биохимии и молекулярной биологии. 51 (4): 282–93. Дои:10.3109/10409238.2016.1172555. ЧВК 4915340. PMID 27098510.
- ^ Камата К., Мицуя М., Нисимура Т., Эйки Дж., Нагата И. (март 2004 г.). «Структурные основы аллостерической регуляции мономерного аллостерического фермента глюкокиназы человека». Структура. 12 (3): 429–38. Дои:10.1016 / j.str.2004.02.005. PMID 15016359.
- ^ Froguel P, Zouali H, Vionnet N, Velho G, Vaxillaire M, Sun F, Lesage S, Stoffel M, Takeda J, Passa P (март 1993 г.). «Семейная гипергликемия из-за мутаций в глюкокиназе. Определение подтипа сахарного диабета». Медицинский журнал Новой Англии. 328 (10): 697–702. Дои:10.1056 / NEJM199303113281005. PMID 8433729.
- ^ Окада С., О'Брайен Дж. С. (август 1969 г.). «Болезнь Тея – Сакса: общее отсутствие компонента бета-D-N-ацетилгексозаминидазы». Наука. 165 (3894): 698–700. Bibcode:1969Sci ... 165..698O. Дои:10.1126 / science.165.3894.698. PMID 5793973. S2CID 8473726.
- ^ "Изучение болезни Тея – Сакса". Национальный институт исследования генома человека США. Получено 1 марта 2015.
- ^ Эрландсен Х., Стивенс Р.С. (октябрь 1999 г.). «Структурные основы фенилкетонурии». Молекулярная генетика и метаболизм. 68 (2): 103–25. Дои:10.1006 / мг.1999.2922. PMID 10527663.
- ^ Flatmark T, Стивенс Р.К. (август 1999 г.). «Структурное понимание гидроксилаз ароматических аминокислот и их мутантных форм, связанных с заболеванием». Химические обзоры. 99 (8): 2137–2160. Дои:10.1021 / cr980450y. PMID 11849022.
- ^ «Фенилкетонурия». Гены и болезни [Интернет]. Bethesda (MD): Национальный центр биотехнологической информации (США). 1998–2015 гг.
- ^ «Недостаток псевдохолинэстеразы». Национальная медицинская библиотека США. Получено 5 сентября 2013.
- ^ Фикер А., Филпотт Дж., Арман М (2011). «Заместительная ферментная терапия недостаточности поджелудочной железы: настоящее и будущее». Клиническая и экспериментальная гастроэнтерология. 4: 55–73. Дои:10.2147 / CEG.S17634. ЧВК 3132852. PMID 21753892.
- ^ Misselwitz B, Pohl D, Frühauf H, Fried M, Vavricka SR, Fox M (июнь 2013 г.). «Мальабсорбция и непереносимость лактозы: патогенез, диагностика и лечение». Единый европейский гастроэнтерологический журнал. 1 (3): 151–9. Дои:10.1177/2050640613484463. ЧВК 4040760. PMID 24917953.
- ^ Кливер Дж. Э. (май 1968 г.). «Дефектная репликация репарации ДНК в пигментной ксеродермии». Природа. 218 (5142): 652–6. Bibcode:1968Натура.218..652С. Дои:10.1038 / 218652a0. PMID 5655953. S2CID 4171859.
- ^ Джеймс В.Д., Элстон Д., Бергер Т.Г. (2011). Болезни кожи Эндрюса: клиническая дерматология (11-е изд.). Лондон: Сондерс / Эльзевир. п. 567. ISBN 978-1437703146.
- ^ Мурзин, А. Г. (1993). «Могут ли гомологичные белки проявлять различную ферментативную активность?». Тенденции в биохимических науках. 18 (11): 403–405. Дои:10.1016/0968-0004(93)90132-7. ISSN 0968-0004. PMID 8291080.
- ^ Очоа, Дэвид; Брэдли, Дэвид; Бельтрао, Педро (2018). «Эволюция, динамика и нарушение регуляции передачи сигналов киназы». Текущее мнение в структурной биологии. 48: 133–140. Дои:10.1016 / j.sbi.2017.12.008. ISSN 1879-033X. PMID 29316484.
- ^ Ренугопалакришнан В., Гардуньо-Хуарес Р., Нарасимхан Г., Верма С.С., Вей Х, Ли П. (ноябрь 2005 г.). «Рациональный дизайн термостабильных белков: актуальность для бионанотехнологии». Журнал нанонауки и нанотехнологий. 5 (11): 1759–1767. Дои:10.1166 / jnn.2005.441. PMID 16433409.
- ^ Халт К., Берглунд П. (август 2003 г.). «Разработанные ферменты для улучшенного органического синтеза». Текущее мнение в области биотехнологии. 14 (4): 395–400. Дои:10.1016 / S0958-1669 (03) 00095-8. PMID 12943848.
- ^ Jiang L, Althoff EA, Clemente FR, Doyle L, Röthlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas CF, Hilvert D, Houk KN, Stoddard BL, Baker D (март 2008 г.). «Вычислительный дизайн ретроальдольных ферментов de novo». Наука. 319 (5868): 1387–91. Bibcode:2008Sci ... 319.1387J. Дои:10.1126 / science.1152692. ЧВК 3431203. PMID 18323453.
- ^ а б Сунь Й., Ченг Дж. (Май 2002 г.). «Гидролиз лигноцеллюлозных материалов для производства этанола: обзор». Биоресурсные технологии. 83 (1): 1–11. Дои:10.1016 / S0960-8524 (01) 00212-7. PMID 12058826.
- ^ а б Кирк О., Borchert TV, Fuglsang CC (август 2002 г.). «Промышленные ферменты». Текущее мнение в области биотехнологии. 13 (4): 345–351. Дои:10.1016 / S0958-1669 (02) 00328-2. PMID 12323357.
- ^ а б c Бриггс Д.Е. (1998). Солод и солод (1-е изд.). Лондон: Блэки Академик. ISBN 978-0412298004.
- ^ Дулье С., Молл М., Будран Дж., Понселе Д. (2000). «Улучшенные характеристики и контроль ферментации пива с помощью инкапсулированной альфа-ацетолактат декарбоксилазы и моделирования». Прогресс биотехнологии. 16 (6): 958–65. Дои:10.1021 / bp000128k. PMID 11101321. S2CID 25674881.
- ^ Tarté R (2008). Ингредиенты в мясных продуктах Свойства, функции и применение. Нью-Йорк: Спрингер. п. 177. ISBN 978-0-387-71327-4.
- ^ «Химозин - База данных ГМО». ГМО Компас. Евросоюз. 10 июля 2010 г. Архивировано с оригинал 26 марта 2015 г.. Получено 1 марта 2015.
- ^ Молимард П., Spinnler HE (февраль 1996 г.). «Обзор: Соединения, участвующие во вкусе сыров, созревших в плесени: происхождение и свойства». Журнал молочной науки. 79 (2): 169–184. Дои:10.3168 / jds.S0022-0302 (96) 76348-8.
- ^ Гусман-Мальдонадо Х., Паредес-Лопес О. (сентябрь 1995 г.). «Амилолитические ферменты и продукты, полученные из крахмала: обзор». Критические обзоры в области пищевой науки и питания. 35 (5): 373–403. Дои:10.1080/10408399509527706. PMID 8573280.
- ^ а б «Протеаза - База данных ГМО». ГМО Компас. Евросоюз. 10 июля 2010 г. Архивировано с оригинал 24 февраля 2015 г.. Получено 28 февраля 2015.
- ^ Алькорта I, Гарбису К., Лама М.Дж., Серра Д.Л. (январь 1998 г.). «Промышленное применение пектиновых ферментов: обзор». Биохимия процесса. 33 (1): 21–28. Дои:10.1016 / S0032-9592 (97) 00046-0.
- ^ Баджпай П. (март 1999 г.). «Применение ферментов в целлюлозно-бумажной промышленности». Прогресс биотехнологии. 15 (2): 147–157. Дои:10.1021 / bp990013k. PMID 10194388. S2CID 26080240.
- ^ Бегли CG, Paragina S, Sporn A (март 1990). «Анализ ферментных очистителей контактных линз». Журнал Американской оптометрической ассоциации. 61 (3): 190–4. PMID 2186082.
- ^ Фаррис П.Л. (2009 г.). «Экономический рост и организация крахмальной промышленности США». В BeMiller JN, Whistler RL (ред.). Химия и технология крахмала (3-е изд.). Лондон: Академ. ISBN 9780080926551.
дальнейшее чтение
Общий
Этимология и история
| Структура и механизм фермента
Кинетика и ингибирование
|