Фенилаланингидроксилаза - Phenylalanine hydroxylase



Фенилаланингидроксилаза (ПАУ) (EC 1.14.16.1 ) является фермент что катализирует гидроксилирование ароматической боковой цепи фенилаланин чтобы генерировать тирозин. ПАУ является одним из трех членов биоптерин -зависимый гидроксилазы ароматических аминокислот, класс монооксигеназа который использует тетрагидробиоптерин (BH4, а птеридин кофактор) и негемовое железо для катализа. В ходе реакции молекулярный кислород гетеролитически расщепляется с последовательным встраиванием одного атома кислорода в BH.4 и фенилаланин-субстрат.[5]
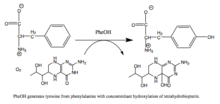 Реакция, катализируемая ПАУ |
Фенилаланингидроксилаза - это ограничение скорости фермент метаболический путь который разрушает избыток фенилаланина. Исследование фенилаланингидроксилазы Сеймуром Кауфманом привело к открытию тетрагидробиоптерина в качестве биологического кофактора.[6] Фермент также интересен с точки зрения здоровья человека, потому что мутации в ПАУ, кодирующий ген, может привести к фенилкетонурия, тяжелое нарушение обмена веществ.
Ферментный механизм
Считается, что реакция проходит через следующие этапы:
- образование Fe (II) -O-O-BH4 мост.
- гетеролитический разрыв связи O-O с образованием промежуточного продукта феррилоксогидроксилирования Fe (IV) = O
- атака на Fe (IV) = O с гидроксилированием фенилаланинового субстрата до тирозина.[7]

Образование и расщепление моста железо-пероксиптерин. Хотя данные убедительно подтверждают, что Fe (IV) = O является промежуточным продуктом гидроксилирования,[8] механистические детали, лежащие в основе образования Fe (II) -O-O-BH4 мостик перед гетеролитическим расщеплением остается спорным. Были предложены два пути, основанные на моделях, которые различаются близостью железа к кофактору птерина и числом молекул воды, которые, как предполагается, координируются железом во время катализа. Согласно одной модели, комплекс железа с диоксидом кислорода первоначально образуется и стабилизируется как резонансный гибрид Fe2+О2 и Fe3+О2−. Активированный O2 затем атакует BH4, образуя переходное состояние, характеризующееся разделением зарядов между электронодефицитным птериновым кольцом и богатыми электронами дикислородом.[9] Впоследствии образуется мостик Fe (II) -O-O-BH4. С другой стороны, формирование этого мостика моделировалось в предположении, что BH4 находится в первой координационной оболочке железа и что железо не координируется ни с какими молекулами воды. Эта модель предсказывает другой механизм с участием радикала птерина и супероксида в качестве критических промежуточных продуктов.[10] После образования Fe (II) -O-O-BH4 мостик разрывается в результате гетеролитического расщепления связи O-O с Fe (IV) = O и 4a-гидрокситетрагидробиоптерином; таким образом, молекулярный кислород является источником обоих атомов кислорода, используемых для гидроксилирования кольца птерина и фенилаланина.
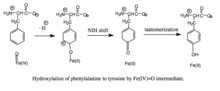
Гидроксилирование фенилаланина промежуточным феррилоксо. Поскольку механизм включает промежуточное соединение гидроксилирования Fe (IV) = O (в отличие от пероксиптерина), окисление BH4 кофактор и гидроксилирование фенилаланина могут быть разделены, что приводит к непродуктивному потреблению BH4 и образованию H2O2.[7] Однако при продуктивности промежуточное соединение Fe (IV) = O добавляется к фенилаланину в реакции электрофильного ароматического замещения, которая восстанавливает железо из феррила в двухвалентное состояние.[7] Хотя первоначально был предложен ареноксид или радикальный промежуточный продукт, анализ родственных триптофана и тирозингидроксилаз показал, что реакция вместо этого протекает через катионный промежуточный продукт, который требует, чтобы Fe (IV) = O координировался с водным лигандом, а не с гидроксогруппой. .[7][11] Этот катионный промежуточный продукт впоследствии подвергается 1,2-гидридному сдвигу NIH, давая промежуточный диенон, который затем таутомеризуется с образованием тирозинового продукта.[12] Кофактор птерина регенерируется путем гидратации карбиноламинового продукта ПАУ до хиноноида дигидробиоптерина (qBH2), которая затем сводится к BH4.[13]
Ферментативная регуляция
ПАУ предлагается использовать морфеин модель аллостерическая регуляция.[14][15]
ПАУ млекопитающих существует в равновесии, состоящем из тетрамеров двух различных архитектур, с одной или несколькими димерными формами как частью равновесия. Такое поведение согласуется с диссоциативным аллостерическим механизмом.[15]
Многие исследования показывают, что ЛАГ млекопитающих демонстрирует поведение, сопоставимое с порфобилиногенсинтаза (PBGS), где, как сообщается, различные факторы, такие как pH и связывание лиганда, влияют на активность фермента и стабильность белка.[15]
Структура
Мономер ПАУ (51,9 кДа) состоит из трех отдельных доменов: регуляторного N-концевого домена (остатки 1–117), который содержит Phe-связывающий субдомен ACT, каталитического домена (остатки 118–427) и C-концевого домена. (остатки 428–453), ответственные за олигомеризацию идентичных мономеров. Был проведен обширный кристаллографический анализ, особенно каталитического домена, координированного птерином и железом, для исследования активного сайта. Также была определена структура N-концевого регуляторного домена, и вместе с решенной структурой гомологичного C-концевого домена тетрамеризации тирозингидроксилазы была предложена структурная модель тетрамерного PAH.[13] Используя рентгеновскую кристаллографию, структура полноразмерного ПАУ крысы была определена экспериментально и показала автоингибированную форму фермента или форму состояния покоя.[16] Форма в состоянии покоя (RS-PAH) архитектурно отличается от активированной формы (A-PAH).[17] Полноразмерная структура A-PAH в настоящее время отсутствует, но был определен стабилизированный Phe интерфейс ACT-ACT, который характерен для A-PAH, и была предложена структурная модель A-PAH, основанная на анализе SAXS.[18][19]
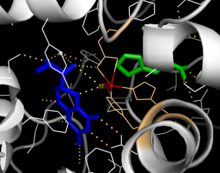
Каталитический домен
Решенные кристаллические структуры каталитического домена показывают, что активный центр состоит из открытого и просторного кармана, выстланного в основном гидрофобными остатками, хотя также присутствуют три остатка глутаминовой кислоты, два гистидина и тирозин, связывающий железо.[13] Существуют противоречивые данные о состоянии координации атома двухвалентного железа и его близости к BH4 в активном центре. Согласно кристаллографическому анализу, Fe (II) координируется водой, His285, His290 и Glu330 (расположение лицевой триады 2-his-1-карбоксилата) с октаэдрической геометрией.[20] Включение аналога Phe в кристаллическую структуру изменяет как железо из шести- в пятикоординированное состояние, включая одиночную молекулу воды, так и бидентатную координацию до Glu330 и открывая место для связывания кислорода. При этом BH4 смещается в сторону атома железа, хотя кофактор птерина остается во второй координационной сфере.[21] С другой стороны, конкурирующая модель, основанная на анализе ЯМР и молекулярного моделирования, предполагает, что все скоординированные молекулы воды вытесняются из активного центра во время каталитического цикла, в то время как BH4 становится напрямую скоординированным с железом.[22] Как обсуждалось выше, устранение этого несоответствия будет важно для определения точного механизма катализа ПАУ.
N-концевой регуляторный домен
Регуляторная природа N-концевого домена (остатки 1–117) обеспечивается его структурной гибкостью.[23] Анализ обменов водород / дейтерий показывает, что аллостерическое связывание Phe глобально изменяет конформацию ПАУ, так что активный центр менее закрывается, поскольку поверхность раздела между регуляторным и каталитическим доменами все больше подвергается воздействию растворителя.[23][24][25] Это наблюдение согласуется с кинетическими исследованиями, которые показывают изначально низкую скорость образования тирозина для полноразмерных ПАУ. Однако это время задержки не наблюдается для усеченного PAH, лишенного N-концевого домена, или если полноразмерный фермент предварительно инкубируют с Phe. Удаление N-концевого домена также устраняет время задержки, увеличивая при этом сродство к Phe почти в два раза; в VМаксимум или Kм для кофактора тетрагидробиоптерина.[26] Дополнительное регулирование обеспечивается Ser16; фосфорилирование этого остатка не изменяет конформацию фермента, но снижает концентрацию Phe, необходимую для аллостерической активации.[25] Этот N-концевой регуляторный домен не наблюдается в бактериальных ПАУ, но демонстрирует значительную структурную гомологию с регуляторным доменом фосфогилцератдегидрогеназы, фермента в пути биосинтеза серина.[25]
Домен тетрамеризации
Прокариотический ПАУ является мономерным, тогда как эукариотический ПАУ существует в равновесии между гомотетрамерными и гомодимерными формами.[7][13] Интерфейс димеризации состоит из петель, связанных с симметрией, которые связывают идентичные мономеры, в то время как перекрывающийся C-концевой домен тетрамеризации опосредует ассоциацию конформационно различных димеров, которые характеризуются различной относительной ориентацией каталитического и тетрамеризационного доменов (Flatmark, Erlandsen). Возникающее в результате искажение симметрии тетрамера проявляется в разной площади поверхности границ раздела димеризации и отличает ПАУ от тетрамерно-симметричной тирозингидроксилазы.[13] Механизм обмена доменов был предложен для опосредования образования тетрамера из димеров, в котором С-концевые альфа-спирали взаимно изменяют свою конформацию вокруг гибкой С-концевой шарнирной области из пяти остатков, образуя структуру спиральной спирали, сдвигая равновесие. в сторону тетрамерной формы.[7][13][27] Хотя как гомодимерные, так и гомотетрамерные формы ПАУ каталитически активны, они демонстрируют дифференциальную кинетику и регуляцию. Помимо пониженной каталитической эффективности, димер не проявляет положительной кооперативности по отношению к L-Phe (который при высоких концентрациях активирует фермент), что позволяет предположить, что L-Phe аллостерически регулирует PAH, влияя на взаимодействие димер-димер.[27]
Биологическая функция
ПАУ - важный фермент в фенилаланин метаболизм и катализирует лимитирующую стадию в его полном катаболизм в углекислый газ и воду.[13][28] Регуляция потока через фенилаланин-ассоциированные пути имеет решающее значение для метаболизма млекопитающих, о чем свидетельствует токсичность высоких уровней этой аминокислоты в плазме крови, наблюдаемых в фенилкетонурия (Смотри ниже). Основным источником фенилаланина являются белки, поступающие с пищей, но относительно небольшая часть этого пула используется для синтеза белка.[28] Вместо этого большая часть попавшего внутрь фенилаланина катаболизируется через ПАУ с образованием тирозин; добавление гидроксильной группы позволяет разорвать бензольное кольцо на последующих катаболических стадиях. Трансаминирование к фенилпируват, метаболиты которого выводятся с мочой, представляет собой другой путь обмена фенилаланина, но преобладает катаболизм через ПАУ.[28]
У людей этот фермент экспрессируется как в печени, так и в почках, и есть некоторые указания на то, что он может по-разному регулироваться в этих тканях.[29] ПАУ необычен среди гидроксилаз ароматических аминокислот из-за его участия в катаболизме; тирозин и триптофангидроксилазы, с другой стороны, в первую очередь экспрессируются в центральной нервной системе и катализируют лимитирующие скорость шаги в биосинтезе нейромедиаторов / гормонов.[13]
Актуальность болезни
Дефицит активности ПАУ из-за мутаций в ПАУ причины гиперфенилаланинемия (HPA), и когда уровень фенилаланина в крови увеличивается более чем в 20 раз по сравнению с нормальной концентрацией, нарушение обмена веществ фенилкетонурия (ФКУ) результаты.[28] ФКУ является как генотипически, так и фенотипически гетерогенным: было идентифицировано более 300 различных патогенных вариантов, большинство из которых соответствуют миссенс-мутациям, которые отображаются в каталитическом домене.[13][20] Когда когорта идентифицированных мутантов PAH экспрессировалась в рекомбинантных системах, ферменты проявляли измененное кинетическое поведение и / или пониженную стабильность, что согласуется со структурным картированием этих мутаций как в каталитическом, так и в тетрамеризационном доменах фермента.[13] BH44 вводился в качестве фармакологического лечения, и было показано, что он снижает уровень фенилаланина в крови у части пациентов с фенилкетонурией, генотипы которых приводят к некоторой остаточной активности ЛАГ, но не имеют дефекта в BH44 синтез или регенерация. Последующие исследования показывают, что в случае некоторых мутантов PAH избыток BH44 действует как фармакологический шаперон для стабилизации мутантных ферментов с нарушенной сборкой тетрамера и повышенной чувствительностью к протеолитическому расщеплению и агрегации.[30] Мутации, которые были идентифицированы в локусе PAH, задокументированы в базе знаний локуса фенилаланингидроксилазы (PAHdb, https://web.archive.org/web/20130718162051/http://www.pahdb.mcgill.ca/ ).
Поскольку фенилкетонурия может вызвать необратимые повреждения, крайне важно, чтобы дефицит фенилаланингидроксилазы определялся на ранней стадии развития. Первоначально это было сделано с использованием анализа ингибирования бактерий, известного как Тест Гатри. Теперь ФКУ является частью обследование новорожденных во многих странах повышенный уровень фенилаланина выявляется вскоре после рождения путем измерения с помощью тандемная масс-спектрометрия. Посадка человека на диету с низким содержанием фенилаланина и высоким содержанием тирозина может помочь предотвратить любой долгосрочный ущерб его развитию.
Родственные ферменты
Фенилаланингидроксилаза тесно связана с двумя другими ферментами:
- триптофангидроксилаза (Номер ЕС 1.14.16.4), который контролирует уровни серотонин в мозгу и желудочно-кишечный тракт
- тирозингидроксилаза (Номер ЕС 1.14.16.2), который контролирует уровни дофамин, адреналин, и норэпинефрин в головном мозге и мозговом веществе надпочечников.
Три фермента гомологичны, то есть, как считается, произошли от одной и той же древней гидроксилазы.
Рекомендации
- ^ а б c ГРЧ38: Ансамбль выпуск 89: ENSG00000171759 - Ансамбль, Май 2017
- ^ а б c GRCm38: выпуск Ensembl 89: ENSMUSG00000020051 - Ансамбль, Май 2017
- ^ "Справочник человека по PubMed:". Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ «Ссылка на Mouse PubMed:». Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ Фитцпатрик П.Ф. (1999). «Тетрагидроптерин-зависимые аминокислотные гидроксилазы». Ежегодный обзор биохимии. 68: 355–81. Дои:10.1146 / annurev.biochem.68.1.355. PMID 10872454.
- ^ Кауфман С (февраль 1958 г.). «Новый кофактор, необходимый для ферментативного превращения фенилаланина в тирозин». Журнал биологической химии. 230 (2): 931–9. PMID 13525410.
- ^ а б c d е ж Фитцпатрик П.Ф. (декабрь 2003 г.). «Механизм гидроксилирования ароматических аминокислот». Биохимия. 42 (48): 14083–91. Дои:10.1021 / bi035656u. ЧВК 1635487. PMID 14640675.
- ^ Панай А.Дж., Ли М., Кребс С., Боллинджер Дж.М., Фицпатрик П.Ф. (март 2011 г.). «Доказательства наличия высокоспиновых форм Fe (IV) в каталитическом цикле бактериальной фенилаланингидроксилазы». Биохимия. 50 (11): 1928–33. Дои:10.1021 / bi1019868. ЧВК 3059337. PMID 21261288.
- ^ Bassan A, Blomberg MR, Siegbahn PE (январь 2003 г.). «Механизм расщепления кислородом в тетрагидробиоптерин-зависимых аминокислотных гидроксилазах». Химия. 9 (1): 106–15. Дои:10.1002 / chem.200390006. PMID 12506369.
- ^ Олссон Э., Мартинес А., Тейген К., Йенсен В.Р. (март 2011 г.). «Образование оксогидроксилирующих частиц железа в каталитическом цикле гидроксилаз ароматических аминокислот». Химия. 17 (13): 3746–58. Дои:10.1002 / chem.201002910. PMID 21351297.
- ^ Bassan A, Blomberg MR, Siegbahn PE (сентябрь 2003 г.). «Механизм ароматического гидроксилирования активированным ядром FeIV = O в тетрагидробиоптерин-зависимых гидроксилазах». Химия. 9 (17): 4055–67. Дои:10.1002 / chem.200304768. PMID 12953191.
- ^ Павон Дж. А., Фицпатрик П. Ф. (сентябрь 2006 г.). «Понимание каталитических механизмов фенилаланина и триптофангидроксилазы на основе кинетических изотопных эффектов на ароматическое гидроксилирование». Биохимия. 45 (36): 11030–7. Дои:10.1021 / bi0607554. ЧВК 1945167. PMID 16953590.
- ^ а б c d е ж грамм час я j Flatmark T, Стивенс Р.С. (август 1999 г.). «Структурное понимание гидроксилаз ароматических аминокислот и их мутантных форм, связанных с заболеванием». Химические обзоры. 99 (8): 2137–2160. Дои:10.1021 / cr980450y. PMID 11849022.
- ^ Селвуд Т., Джаффе Е.К. (март 2012 г.). «Динамические диссоциирующие гомоолигомеры и контроль функции белка». Архивы биохимии и биофизики. 519 (2): 131–43. Дои:10.1016 / j.abb.2011.11.020. ЧВК 3298769. PMID 22182754.
- ^ а б c Джаффе Е.К., Стит Л., Лоуренс С.Х., Андрейк М., Данбрак Р.Л. (февраль 2013 г.). «Новая модель аллостерической регуляции фенилаланингидроксилазы: последствия для болезней и терапии». Архивы биохимии и биофизики. 530 (2): 73–82. Дои:10.1016 / j.abb.2012.12.017. ЧВК 3580015. PMID 23296088.
- ^ Артуро Е.К., Гупта К., Эру А., Стит Л., Кросс П.Дж., Паркер Е.Дж., Лолл П.Дж., Яффе Е.К. (март 2016 г.). «Первая структура полноразмерной фенилаланингидроксилазы млекопитающих раскрывает архитектуру аутоингибированного тетрамера». Труды Национальной академии наук Соединенных Штатов Америки. 113 (9): 2394–9. Дои:10.1073 / pnas.1516967113. ЧВК 4780608. PMID 26884182.
- ^ Jaffe EK (август 2017 г.). «Новые белковые структуры обеспечивают обновленное понимание фенилкетонурии». Молекулярная генетика и метаболизм. 121 (4): 289–296. Дои:10.1016 / j.ymgme.2017.06.005. ЧВК 5549558. PMID 28645531.
- ^ Патель Д., Копек Дж., Фицпатрик Ф., МакКорви Т.Дж., Юэ В.В. (апрель 2016 г.). «Структурные основы лиганд-зависимой димеризации регуляторного домена фенилаланингидроксилазы». Научные отчеты. 6 (1): 23748. Дои:10.1038 / srep23748. ЧВК 4822156. PMID 27049649.
- ^ Meisburger SP, Taylor AB, Khan CA, Zhang S, Fitzpatrick PF, Ando N (май 2016 г.). «Движение доменов при активации фенилаланингидроксилазы, характеризуемое кристаллографией и малоугловым рентгеновским рассеянием в сочетании с хроматографией». Журнал Американского химического общества. 138 (20): 6506–16. Дои:10.1021 / jacs.6b01563. ЧВК 4896396. PMID 27145334.
- ^ а б Эрландсен Х., Фузетти Ф., Мартинес А., Хаф Э., Флэтмарк Т., Стивенс Р.С. (декабрь 1997 г.). «Кристаллическая структура каталитического домена фенилаланингидроксилазы человека раскрывает структурную основу фенилкетонурии». Структурная биология природы. 4 (12): 995–1000. Дои:10.1038 / nsb1297-995. PMID 9406548. S2CID 6293946.
- ^ Андерсен О.А., Флэтмарк Т., Хаф Э. (июль 2002 г.). «Кристаллическая структура тройного комплекса каталитического домена фенилаланингидроксилазы человека с тетрагидробиоптерином и 3- (2-тиенил) -L-аланином, и ее значение для механизма катализа и активации субстрата». Журнал молекулярной биологии. 320 (5): 1095–108. Дои:10.1016 / S0022-2836 (02) 00560-0. PMID 12126628.
- ^ Тейген К., Фрёйштейн Н.А., Мартинес А. (декабрь 1999 г.). «Структурная основа распознавания кофакторов фенилаланина и птерина фенилаланингидроксилазой: последствия для каталитического механизма». Журнал молекулярной биологии. 294 (3): 807–23. Дои:10.1006 / jmbi.1999.3288. PMID 10610798.
- ^ а б Ли Дж., Данготт Л.Дж., Фицпатрик П.Ф. (апрель 2010 г.). «Регулирование фенилаланингидроксилазы: конформационные изменения при связывании фенилаланина, обнаруженные с помощью обмена водорода / дейтерия и масс-спектрометрии». Биохимия. 49 (15): 3327–35. Дои:10.1021 / bi1001294. ЧВК 2855537. PMID 20307070.
- ^ Ли Дж., Илангован Ю., Даубнер С.К., Хинк А.П., Фитцпатрик П.Ф. (январь 2011 г.). «Прямые доказательства наличия фенилаланинового сайта в регуляторном домене фенилаланингидроксилазы». Архивы биохимии и биофизики. 505 (2): 250–5. Дои:10.1016 / j.abb.2010.10.009. ЧВК 3019263. PMID 20951114.
- ^ а б c Коби Б., Дженнингс И.Г., Хаус К.М., Мичелл Б.Дж., Гудвилл К.Э., Сантарсиеро Б.Д., Стивенс Р.С., Коттон Р.Г., Кемп Б.Э. (май 1999 г.). «Структурные основы ауторегуляции фенилаланингидроксилазы». Структурная биология природы. 6 (5): 442–8. Дои:10.1038/8247. PMID 10331871. S2CID 11709986.
- ^ Daubner SC, Hillas PJ, Fitzpatrick PF (декабрь 1997 г.). «Экспрессия и характеристика каталитического домена фенилаланингидроксилазы человека». Архивы биохимии и биофизики. 348 (2): 295–302. Дои:10.1006 / abbi.1997.0435. PMID 9434741.
- ^ а б Bjørgo E, de Carvalho RM, Flatmark T (февраль 2001 г.). «Сравнение кинетических и регуляторных свойств тетрамерных и димерных форм дикого типа и мутантной Thr427 -> Pro фенилаланингидроксилазы человека: вклад гибкой шарнирной области Asp425-Gln429 в тетрамеризацию и кооперативное связывание субстрата». Европейский журнал биохимии. 268 (4): 997–1005. Дои:10.1046 / j.1432-1327.2001.01958.x. PMID 11179966.
- ^ а б c d Кауфман С (март 1999 г.). «Модель метаболизма фенилаланина человека у здоровых людей и пациентов с фенилкетонурией». Труды Национальной академии наук Соединенных Штатов Америки. 96 (6): 3160–4. Дои:10.1073 / пнас.96.6.3160. ЧВК 15912. PMID 10077654.
- ^ Lichter-Konecki U, Hipke CM, Konecki DS (август 1999 г.). «Экспрессия гена фенилаланингидроксилазы человека в почках и других непеченочных тканях». Молекулярная генетика и метаболизм. 67 (4): 308–16. Дои:10.1006 / мг.1999.2880. PMID 10444341.
- ^ Muntau AC, Gersting SW (декабрь 2010 г.). «Фенилкетонурия как модель заболеваний неправильного сворачивания белков и для разработки орфанных препаратов следующего поколения для пациентов с врожденными нарушениями метаболизма». Журнал наследственных метаболических заболеваний. 33 (6): 649–58. Дои:10.1007 / s10545-010-9185-4. PMID 20824346. S2CID 20843095.
дальнейшее чтение
- Eisensmith RC, Woo SL (1993). «Молекулярные основы фенилкетонурии и родственных гиперфенилаланинемий: мутации и полиморфизмы в гене фенилаланингидроксилазы человека». Человеческая мутация. 1 (1): 13–23. Дои:10.1002 / humu.1380010104. PMID 1301187. S2CID 19476605.
- Konecki DS, Lichter-Konecki U (август 1991 г.). «Локус фенилкетонурии: современные знания об аллелях и мутациях гена фенилаланингидроксилазы в различных популяциях». Генетика человека. 87 (4): 377–88. Дои:10.1007 / BF00197152. PMID 1679029. S2CID 25627287.
- Коттон Р.Г. (1991). «Гетерогенность фенилкетонурии на клиническом, белковом и ДНК уровне». Журнал наследственных метаболических заболеваний. 13 (5): 739–50. Дои:10.1007 / BF01799577. PMID 2246858. S2CID 21931016.
- Эрландсен Х., Фузетти Ф., Мартинес А., Хаф Э., Флэтмарк Т., Стивенс Р.С. (декабрь 1997 г.). «Кристаллическая структура каталитического домена фенилаланингидроксилазы человека раскрывает структурную основу фенилкетонурии». Структурная биология природы. 4 (12): 995–1000. Дои:10.1038 / nsb1297-995. PMID 9406548. S2CID 6293946.
- Waters PJ, Parniak MA, Nowacki P, Scriver CR (1998). «Анализ экспрессии мутаций в фенилаланингидроксилазе in vitro: связь генотипа с фенотипом и структуры с функцией». Человеческая мутация. 11 (1): 4–17. Дои:10.1002 / (SICI) 1098-1004 (1998) 11: 1 <4 :: AID-HUMU2> 3.0.CO; 2-L. PMID 9450897.
- Waters PJ (апрель 2003 г.). «Как мутации гена PAH вызывают гиперфенилаланинемию и почему механизм имеет значение: выводы из экспрессии in vitro». Человеческая мутация. 21 (4): 357–69. Дои:10.1002 / humu.10197. PMID 12655545. S2CID 23769500.
внешняя ссылка
- GeneReviews / NCBI / NIH / UW запись о дефиците фенилаланингидроксилазы
- Локус-специфическая база данных вариантов гена фенилаланингидроксилазы человека
- Молекула месяца: фенилаланингидроксилаза
- Обзор всей структурной информации, доступной в PDB за UniProt: P00439 (Фенилаланингидроксилаза человека) на PDBe-KB.
PDB галерея | |
|---|---|
|