Тирозин аминотрансфераза - Tyrosine aminotransferase
| Тирозин трансаминаза | |||||||||
|---|---|---|---|---|---|---|---|---|---|
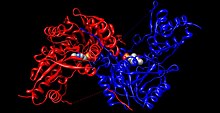
 Человеческая тирозинаминотрансфераза (цвет радуги, N-конец = синий, C-конец = красный) в комплексе с пиридоксальфосфат (модель, заполняющая пространство ).[1] | |||||||||
| Идентификаторы | |||||||||
| Номер ЕС | 2.6.1.5 | ||||||||
| Количество CAS | 9014-55-5 | ||||||||
| Базы данных | |||||||||
| IntEnz | Просмотр IntEnz | ||||||||
| БРЕНДА | BRENDA запись | ||||||||
| ExPASy | Просмотр NiceZyme | ||||||||
| КЕГГ | Запись в KEGG | ||||||||
| MetaCyc | метаболический путь | ||||||||
| ПРИАМ | профиль | ||||||||
| PDB структуры | RCSB PDB PDBe PDBsum | ||||||||
| Генная онтология | AmiGO / QuickGO | ||||||||
| |||||||||




Тирозин аминотрансфераза (или же тирозинтрансаминаза) представляет собой фермент, присутствующий в печени и катализирующий превращение тирозин к 4-гидроксифенилпируват.[6]


У человека белок тирозинаминотрансферазы кодируется ТАТ ген.[7] Дефицит фермента у людей может привести к так называемому: тирозинемия типа II, где тирозин в избытке является результатом того, что тирозин не проходит аминотрансферазную реакцию с образованием 4-гидроксифенилпирувата.[8]
Механизм
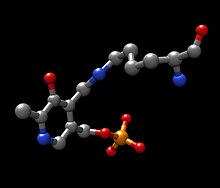
Структуры трех основных молекул, участвующих в химической реакции, катализируемой ферментом тирозинаминотрансферазы, показаны ниже: аминокислота тирозин, то протезная группа пиридоксальфосфат, а полученный продукт 4-гидроксифенилпируват.

Каждая сторона димерного белка включает пиридоксальфосфат (PLP), связанный с Lys280 остаток молекулы тирозинаминотрансферазы. Аминогруппа тирозина атакует альфа-углерод имина, связанного с Lys280, образуя тетраэдрический комплекс, а затем запускает LYS-ENZ. Этот процесс известен как трансмиссия путем отключения иминной группы, связанной с PLP. Затем вновь образованная молекула PLP-TYR подвергается атаке основания.
Возможным кандидатом на основание в механизме может быть Lys280, который только что оттолкнулся от PLP, который секвестрирует вновь образовавшуюся аминогруппу молекулы PLP-TYR. В аналогичном механизме аспартат трансаминаза, лизин, который образует исходный имин для PLP, позже действует как основание, которое атакует тирозин в процессе трансмиссии. Электроны, оставшиеся после потери протона, движутся вниз, чтобы сформировать новую двойную связь с имином, который, в свою очередь, проталкивает электроны с уже двойной связью через PLP и в конечном итоге превращается в неподеленную пару на положительно заряженном азоте в шестичленном атоме азота. кольцо молекулы. Вода атакует альфа-углерод имина PLP-TYR и через ацильное замещение запускает азот PLP с образованием пиридоксаминфосфата (PMP) и 4-гидроксифенилпирувата.
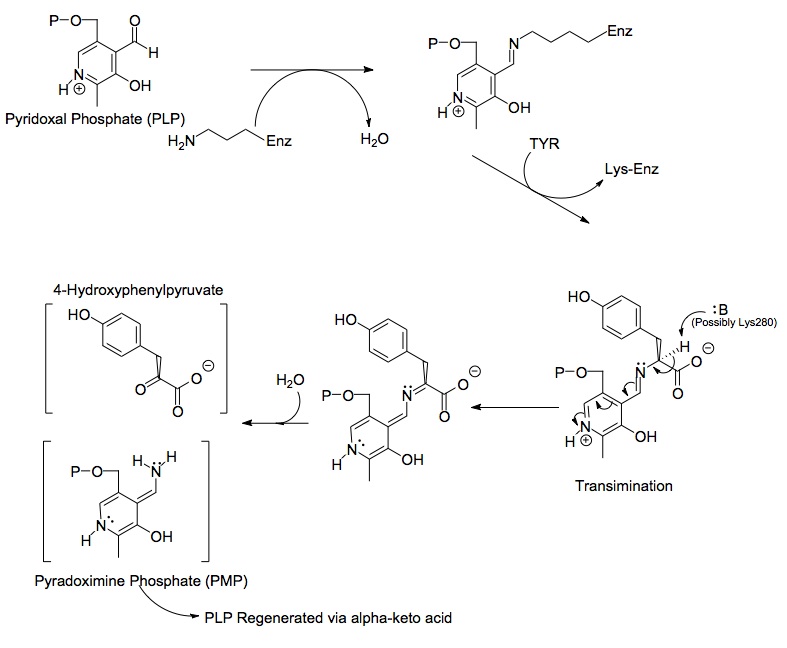
Затем PMP регенерируется в PLP путем переноса своей аминогруппы на альфа-кетоглутарат, реформируя его функциональную альдегидную группу. За этим следует другая реакция замещения остатком Lys280 для преобразования его иминной связи с ферментом с образованием ENZ-PLP.
Активный сайт

Тирозинаминотрансфераза в виде димера имеет два идентичных активных сайта. Lys280 присоединяется к PLP, который удерживается на месте двумя боковыми цепями неполярных аминокислот; фенилаланин и изолейцин (см. эскиз справа). PLP также удерживается на месте за счет водородных связей с окружающими молекулами, в основном за счет своей фосфатной группы.
Ниже показан один активный участок при трех разных увеличениях:
Патология
Тирозинемия является наиболее частым метаболическим заболеванием, связанным с тирозинаминотрансферазой. Заболевание возникает из-за недостаточности печеночной тирозинаминотрансферазы.[10] Тирозинемия II типа (синдром Ричнера-Ханхарта, RHS) - заболевание с аутосомно-рецессивным наследованием, которое характеризуется кератитом, ладонно-подошвенным гиперкератозом, умственной отсталостью и повышенным уровнем тирозина в крови.[10] Кератит у пациентов с тирозинемией II типа вызывается отложением кристаллов тирозина в роговице и приводит к воспалению роговицы.[11] Ген TAT расположен на хромосоме человека 16q22-24 и простирается на 10,9 килобаз (т.п.н.), содержащих 12 экзонов, а его мРНК в 3,0 т.п.н. кодирует белок из 454 аминокислот размером 50,4 кДа.[12] Сообщалось о двенадцати различных мутациях гена ТАТ.[12]
Рекомендации
- ^ а б PDB: 3DYD; Карлберг Т., Моче М., Андерссон Дж. И др. (2008). «Тирозинаминотрансфераза человека». Будут опубликованы.
- ^ а б c ГРЧ38: Ансамбль выпуск 89: ENSG00000198650 - Ансамбль, Май 2017
- ^ а б c GRCm38: выпуск ансамбля 89: ENSMUSG00000001670 - Ансамбль, Май 2017
- ^ "Справочник человека по PubMed:". Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ "Ссылка на Mouse PubMed:". Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ Дитрих Дж. Б. (апрель 1992 г.). «Тирозинаминотрансфераза: трансаминаза среди других?». Клеточная и молекулярная биология. 38 (2): 95–114. PMID 1349265.
- ^ Зеа-Рей, Александра В .; Крус-Камино, Эктор; Васкес-Канту, Диана Л .; Gutiérrez-García, Valeria M .; Сантос-Гусман, Хесус; Канту-Рейна, Консуэло (27 ноября 2017 г.). «Заболеваемость преходящей неонатальной тирозинемией среди населения Мексики». Журнал врожденных ошибок метаболизма и скрининга. 5: 232640981774423. Дои:10.1177/2326409817744230.
- ^ Реттенмайер Р., Натт Э., Зентграф Х., Шерер Г. (июль 1990 г.). «Выделение и характеристика гена тирозинаминотрансферазы человека». Нуклеиновые кислоты Res. 18 (13): 3853–61. Дои:10.1093 / nar / 18.13.3853. ЧВК 331086. PMID 1973834.
- ^ Петтерсен, E.F .; Годдард, Т.Д .; Huang, C.C .; Couch, G.S .; Greenblatt, D.M .; Meng, E.C .; Феррин, Т. (2004). «UCSF Chimera - система визуализации для поисковых исследований и анализа». Журнал вычислительной химии. 25 (13): 1605–1612. CiteSeerX 10.1.1.456.9442. Дои:10.1002 / jcc.20084. PMID 15264254. S2CID 8747218.
- ^ а б Натт Э., Кида К., Одиевр М., Ди Рокко М., Шерер Г. (октябрь 1992 г.). «Точечные мутации в гене тирозинаминотрансферазы при тирозинемии II типа». Proc. Natl. Акад. Sci. СОЕДИНЕННЫЕ ШТАТЫ АМЕРИКИ. 89 (19): 9297–301. Bibcode:1992PNAS ... 89.9297N. Дои:10.1073 / пнас.89.19.9297. ЧВК 50113. PMID 1357662.
- ^ аль-Хемидан А.И., аль-Хазза С.А. (март 1995 г.). «Синдром Ричнера-Ханхарта (тип тирозинемии II). История болезни и обзор литературы». Офтальмологический генет. 16 (1): 21–6. Дои:10.3109/13816819509057850. PMID 7648039.
- ^ а б Минами-Хори М., Исида-Ямамото А., Като Н., Такахаши Х., Иидзука Х. (январь 2006 г.). «Синдром Ричнера-Ханхарта: сообщение о случае с новой мутацией тирозинаминотрансферазы». J. Dermatol. Наука. 41 (1): 82–4. Дои:10.1016 / j.jdermsci.2005.10.007. PMID 16318910.
Молекулярные графические изображения были получены с использованием пакета UCSF Chimera из Ресурса для биокомпьютеров, визуализации и информатики Калифорнийского университета в Сан-Франциско (при поддержке NIH P41 RR-01081).
внешняя ссылка
- Тирозин + аминотрансфераза в Национальной медицинской библиотеке США Рубрики медицинской тематики (MeSH)
- PDBe-KB предоставляет обзор всей структурной информации, доступной в PDB для тирозинаминотрансферазы человека