Галактозо-1-фосфат уридилилтрансфераза - Galactose-1-phosphate uridylyltransferase





| Галактозо-1-фосфатуридилтрансфераза, N-концевой домен | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Идентификаторы | |||||||||
| Символ | GalP_UDP_transf | ||||||||
| Pfam | PF01087 | ||||||||
| Pfam клан | CL0265 | ||||||||
| PROSITE | PDOC00108 | ||||||||
| SCOP2 | 1hxp / Объем / СУПФАМ | ||||||||
| |||||||||
| Галактозо-1-фосфатуридилтрансфераза, С-концевой домен | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 структура нуклеотидилтрансферазы в комплексе с udp-галактозой | |||||||||
| Идентификаторы | |||||||||
| Символ | GalP_UDP_tr_C | ||||||||
| Pfam | PF02744 | ||||||||
| Pfam клан | CL0265 | ||||||||
| ИнтерПро | IPR005850 | ||||||||
| PROSITE | PDOC00108 | ||||||||
| SCOP2 | 1hxp / Объем / СУПФАМ | ||||||||
| |||||||||
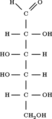
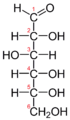
Галактозо-1-фосфат уридилилтрансфераза (или ГАЛТ) является фермент (EC 2.7.7.12 ) отвечает за преобразование полученного галактоза к глюкоза.[5]
Галактозо-1-фосфатуридиллилтрансфераза (GALT) катализирует вторую стадию Leloir путь галактоза обмен веществ, а именно:

Выражение GALT контролируется действиями FOXO3 ген. Отсутствие этого фермента приводит к классической галактоземии у человека и может быть фатальным в период новорожденности, если лактоза не удаляется из рациона. Патофизиология галактоземии четко не определена.[5]
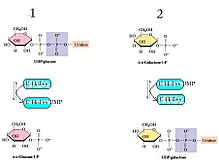
Механизм
GALT катализирует вторую реакцию пути Лелуара метаболизма галактозы через би-би для пинг-понга. кинетика с двойное перемещение механизм.[6] Это означает, что чистая реакция состоит из двух реагентов и двух продуктов (см. Реакцию выше) и протекает по следующему механизму: фермент реагирует с одним субстратом с образованием одного продукта и модифицированного фермента, который продолжает реагировать со вторым. субстрат для получения второго продукта при регенерации исходного фермента.[7] В случае GALT остаток His166 действует как мощный нуклеофил для облегчения переноса нуклеотида между UDP-гексозами и гексозо-1-фосфатами.[8]
- UDP-глюкоза + E-His ⇌ глюкозо-1-фосфат + E-His-UMP
- Галактоза-1-фосфат + E-His-UMP ⇌ UDP-галактоза + E-His[8]
Структурные исследования
Трехмерная структура на 180 вечера разрешающая способность (рентгеновская кристаллография ) GALT был определен Ведекиндом, Фреем и Реймент, и их структурный анализ показал ключевые аминокислоты необходим для функции GALT.[8] Среди них Leu4, Phe75, Asn77, Asp78, Phe79 и Val108, которые согласуются с остатками, которые участвовали как в экспериментах по точечной мутации, так и в клиническом скрининге, которые играют роль в галактоземии человека.[8][10]
Клиническое значение
Причины дефицита GALT классическая галактоземия. Галактоземия является аутосомно-рецессивным наследственным заболеванием, выявляемым у новорожденных и детей.[11] Это происходит примерно у 1 из 40 000-60 000 живорожденных младенцев. Классическая галактоземия (G / G) вызвано дефицитом активности GALT, тогда как более частые клинические проявления, Дуарте (D / D) и Дуарте / классический вариант (D / G) вызваны ослаблением активности GALT.[12] Симптомы включают недостаточность функции яичников, диспраксия (трудно говорить правильно и последовательно),[13] и неврологический дефицит.[12] Единственная мутация в любой из нескольких пар оснований может привести к дефициту активности GALT.[14] Например, одна мутация от A до G в экзоне 6 гена GALT изменяет Glu188 на аргинин а мутация от A до G в экзоне 10 превращает Asn314 в аспарагиновая кислота.[12] Эти две мутации также добавляют новые рестрикционный фермент сайты cut, которые позволяют обнаруживать и проводить крупномасштабный скрининг населения с помощью ПЦР (полимеразной цепной реакции ).[12] Скрининг в основном устранил неонатальную смерть от G / G-галактоземии, но болезнь из-за роли GALT в биохимической метаболизм проглоченного галактоза (который токсичен при накоплении) на энергетически полезный глюкоза, безусловно, может быть фатальным.[11][15] Тем не менее, люди, страдающие галактоземией, могут жить относительно нормальной жизнью, избегая молочных продуктов и всего, что содержит галактозу (потому что она не может быть метаболизирована), но все же существует вероятность проблем с неврологическим развитием или других осложнений, даже у тех, кто избегает галактозы.[16]
База данных болезней
База данных мутаций галактоземии (GALT)
использованная литература
- ^ а б c ГРЧ38: Ансамбль выпуск 89: ENSG00000213930 - Ансамбль, Май 2017
- ^ а б c GRCm38: выпуск Ensembl 89: ENSMUSG00000036073 - Ансамбль, Май 2017
- ^ "Справочник человека по PubMed:". Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ «Ссылка на Mouse PubMed:». Национальный центр биотехнологической информации, Национальная медицинская библиотека США.
- ^ а б «Энтрез Ген: Галактозо-1-фосфатуридилтрансфераза GALT».
- ^ Вонг LJ, Фрей PA (сентябрь 1974 г.). «Галактозо-1-фосфат уридилилтрансфераза: исследования скорости, подтверждающие наличие промежуточного уридилил-фермента на каталитическом пути». Биохимия. 13 (19): 3889–94. Дои:10.1021 / bi00716a011. PMID 4606575.
- ^ «Архивная копия». Архивировано из оригинал на 2016-03-03. Получено 2010-05-19.CS1 maint: заархивированная копия как заголовок (ссылка на сайт)
- ^ а б c d Ведекинд Дж. Э., Фрей П. А., Реймент I (сентябрь 1995 г.). «Трехмерная структура галактозо-1-фосфат уридилилтрансферазы из Escherichia coli при разрешении 1,8 A». Биохимия. 34 (35): 11049–61. Дои:10.1021 / bi00035a010. PMID 7669762.
- ^ «Архивная копия». Архивировано из оригинал на 2008-12-04. Получено 2010-05-19.CS1 maint: заархивированная копия как заголовок (ссылка на сайт)
- ^ Сейрантепе В., Озгуц М., Джошкун Т., Озалп И., Райхардт Дж. К. (1999). «Идентификация мутаций в гене галактозо-1-фосфатуридилтрансферазы (GALT) у 16 турецких пациентов с галактоземией, включая новую мутацию F294Y. Краткое описание мутации № 235. Онлайн». Человеческая мутация. 13 (4): 339. Дои:10.1002 / (SICI) 1098-1004 (1999) 13: 4 <339 :: AID-HUMU18> 3.0.CO; 2-S. PMID 10220154.
- ^ а б Фридович-Кейл Ю.Л. (декабрь 2006 г.). «Галактоземия: хорошее, плохое и неизвестное». Журнал клеточной физиологии. 209 (3): 701–5. Дои:10.1002 / jcp.20820. PMID 17001680. S2CID 32233614.
- ^ а б c d Эльзас Л.Дж., Лэнгли С., Полк Е.М., Хьельм Л.Н., Дембур П.П. (1995). «Молекулярный подход к галактоземии». Европейский журнал педиатрии. 154 (7 Прил. 2): S21-7. Дои:10.1007 / BF02143798. PMID 7671959. S2CID 11937698.
- ^ «Архивная копия». Архивировано из оригинал на 2006-02-28. Получено 2010-05-19.CS1 maint: заархивированная копия как заголовок (ссылка на сайт)
- ^ Добровольски С.Ф., Банас Р.А., Сузов Дж. Г., Беркли М., Нейлор Е. В. (февраль 2003 г.). «Анализ распространенных мутаций в гене галактозо-1-фосфатуридилтрансферазы: новые методы для повышения чувствительности и специфичности скрининга новорожденных на галактоземию». Журнал молекулярной диагностики. 5 (1): 42–7. Дои:10.1016 / S1525-1578 (10) 60450-3. ЧВК 1907369. PMID 12552079.
- ^ Лай К., Эльзас Л.Дж., Вьеренга К.Дж. (ноябрь 2009 г.). «Галактозная токсичность у животных». IUBMB Life. 61 (11): 1063–74. Дои:10.1002 / iub.262. ЧВК 2788023. PMID 19859980.
- ^ http://www.umm.edu/ency/article/000366trt.htm
дальнейшее чтение
- Райхардт Дж. К. (1993). «Генетические основы галактоземии». Человеческая мутация. 1 (3): 190–6. Дои:10.1002 / humu.1380010303. PMID 1301925. S2CID 504197.
- Тайфилд Л., Райхард Дж., Фридович-Кейл Дж., Крок Д. Т., Эльзас Л. Дж., Штробл В., Козак Л., Коскун Т., Новелли Дж., Окано Ю., Зекановски С., Шин И., Боледа М. Д. (1999). «Классическая галактоземия и мутации в гене галактозо-1-фосфат уридилтрансферазы (GALT)». Человеческая мутация. 13 (6): 417–30. Дои:10.1002 / (SICI) 1098-1004 (1999) 13: 6 <417 :: AID-HUMU1> 3.0.CO; 2-0. PMID 10408771.
- Райхардт Дж. К., Белмонт Дж. В., Леви Х. Л., Ву С. Л. (март 1992 г.). «Характеристика двух миссенс-мутаций в человеческой галактозо-1-фосфат уридилтрансферазе: различные молекулярные механизмы галактоземии». Геномика. 12 (3): 596–600. Дои:10.1016 / 0888-7543 (92) 90453-У. PMID 1373122.
- Лесли Н.Д., Иммерман Э.Б., Флак Дж. Э., Флорез М., Фридович-Кейл Дж. Л., Эльзас Л. Дж. (Октябрь 1992 г.). «Ген галактозо-1-фосфат уридилтрансферазы человека». Геномика. 14 (2): 474–80. Дои:10.1016 / S0888-7543 (05) 80244-7. PMID 1427861.
- Райхардт Дж. К., Леви Х. Л., Ву С. Л. (июнь 1992 г.). «Молекулярная характеристика двух мутаций галактоземии и одного полиморфизма: значение для структурно-функционального анализа человеческой галактозо-1-фосфат уридилтрансферазы». Биохимия. 31 (24): 5430–3. Дои:10.1021 / bi00139a002. PMID 1610789.
- Райхардт JK, Packman S, Woo SL (октябрь 1991 г.). «Молекулярная характеристика двух мутаций галактоземии: корреляция мутаций с высококонсервативными доменами в галактозо-1-фосфатуридилтрансферазе». Американский журнал генетики человека. 49 (4): 860–7. ЧВК 1683190. PMID 1897530.
- Райхардт Дж. К., Ву С. Л. (апрель 1991 г.). «Молекулярные основы галактоземии: мутации и полиморфизмы в гене, кодирующем человеческую галактозо-1-фосфатуридиллилтрансферазу». Труды Национальной академии наук Соединенных Штатов Америки. 88 (7): 2633–7. Bibcode:1991ПНАС ... 88.2633R. Дои:10.1073 / пнас.88.7.2633. ЧВК 51292. PMID 2011574.
- Flach JE, Reichardt JK, Elsas LJ (август 1990 г.). «Последовательность кДНК, кодирующая человеческую галактозо-1-фосфатуридилтрансферазу». Молекулярная биология и медицина. 7 (4): 365–9. PMID 2233247.
- Райхардт Дж. К., Берг П. (апрель 1988 г.). «Клонирование и характеристика кДНК, кодирующей человеческую галактозо-1-фосфатуридилтрансферазу». Молекулярная биология и медицина. 5 (2): 107–22. PMID 2840550.
- Бергрен В.Г., Доннелл Г.Н. (июль 1973 г.). «Новый вариант галактозо-1-фосфат уридилтрансферазы в человеке: вариант Лос-Анджелеса». Анналы генетики человека. 37 (1): 1–8. Дои:10.1111 / j.1469-1809.1973.tb01808.x. PMID 4759900. S2CID 22699183.
- Ши Л.Ю., Суслак Л., Розин И., Сирл Б.М., Деспозито Ф. (ноябрь 1984 г.). «Исследования дозировки генов, поддерживающие локализацию структурного гена галактозо-1-фосфатуридилтрансферазы (GALT) в полосе p13 хромосомы 9». Американский журнал медицинской генетики. 19 (3): 539–43. Дои:10.1002 / ajmg.1320190316. PMID 6095663.
- Асино Дж., Окано Й., Суяма И., Ямадзаки Т., Йошино М., Фуруяма Дж., Лин Х. К., Райхардт Дж. К., Иссики Дж. (1995). «Молекулярная характеристика мутаций галактоземии (тип 1) в японском языке». Человеческая мутация. 6 (1): 36–43. Дои:10.1002 / humu.1380060108. PMID 7550229. S2CID 23500152.
- Эльзас Л.Дж., Лэнгли С., Полк Е.М., Хьельм Л.Н., Дембур П.П. (1995). «Молекулярный подход к галактоземии». Европейский журнал педиатрии. 154 (7 Прил. 2): S21-7. Дои:10.1007 / BF02143798. PMID 7671959. S2CID 11937698.
- Эльзас Л.Дж., Лэнгли С., Стил Э., Эвинджер Дж., Фридович-Кейл Д.Л., Браун А., Сингх Р., Фернхофф П., Хьелм Л.Н., Дембур П.П. (март 1995 г.). «Галактоземия: стратегия выявления новых биохимических фенотипов и молекулярных генотипов». Американский журнал генетики человека. 56 (3): 630–9. ЧВК 1801164. PMID 7887416.
- Фридович-Кейл Дж. Л., Лэнгли С. Д., Мазур Л. А., Леннон Дж. К., Дембур П. П., Эльзас Дж. Л. (март 1995 г.). «Идентификация и функциональный анализ трех различных мутаций в гене галактозо-1-фосфат уридилтрансферазы человека, связанных с галактоземией в одной семье». Американский журнал генетики человека. 56 (3): 640–6. ЧВК 1801186. PMID 7887417.
- Давит-Спраул А., Пурси М.Л., Нг К.Х., Сони Т., Лемонье А. (ноябрь 1994 г.). «Регулирующие эффекты галактозы на активность галактозо-1-фосфат уридилтрансферазы на клетках гепатобластомы человека HepG2». Письма FEBS. 354 (2): 232–6. Дои:10.1016/0014-5793(94)01133-8. PMID 7957929. S2CID 45242645.
- Лин ХК, Кирби Л.Т., Нг РГ, Райхардт Дж. К. (февраль 1994 г.). «О молекулярной природе варианта Дуарте галактозо-1-фосфат уридилтрансферазы (GALT)». Генетика человека. 93 (2): 167–9. Дои:10.1007 / BF00210604. PMID 8112740. S2CID 42558872.
- Эльзас Л.Дж., Дембуре П.П., Лэнгли С., Полк Е.М., Хьельм Л.Н., Фридович-Кейл Дж. (Июнь 1994 г.). «Распространенная мутация, связанная с аллелем галактоземии Дуарте». Американский журнал генетики человека. 54 (6): 1030–6. ЧВК 1918187. PMID 8198125.
- Райхардт Дж. К., Новелли Г., Даллапиккола Б. (март 1993 г.). «Молекулярная характеристика мутации галактоземии H319Q». Молекулярная генетика человека. 2 (3): 325–6. Дои:10,1093 / hmg / 2.3.325. PMID 8499924.
внешние ссылки
- Галактоза-1-P-уридилтрансфераза в Национальной медицинской библиотеке США Рубрики медицинской тематики (MeSH)
- GeneReviews / NIH / NCBI / UW запись о галактоземии
- База данных мутаций галактоземии (GALT)
- База данных GALT Protein
- PDBe-KB предоставляет обзор всей структурной информации, доступной в PDB для человеческой галактозо-1-фосфат уридилилтрансферазы