PDE1 - PDE1
| Фосфодиэстераза I | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Идентификаторы | |||||||||
| Номер ЕС | 3.1.4.1 | ||||||||
| Количество CAS | 9025-82-5 | ||||||||
| Базы данных | |||||||||
| IntEnz | Просмотр IntEnz | ||||||||
| БРЕНДА | BRENDA запись | ||||||||
| ExPASy | Просмотр NiceZyme | ||||||||
| КЕГГ | Запись в KEGG | ||||||||
| MetaCyc | метаболический путь | ||||||||
| ПРИАМ | профиль | ||||||||
| PDB структуры | RCSB PDB PDBe PDBsum | ||||||||
| |||||||||
PDE1 (фосфодиэстераза 1 типа) это фосфодиэстераза фермент также известен как кальций - и кальмодулин -зависимая фосфодиэстераза. Это одно из 11 семейств фосфодиэстераз (PDE1-PDE11). PDE1 имеет три подтипы, PDE1A, PDE1B и PDE1C, которые далее делятся на различные изоформы. Различные изоформы проявляют различное сродство к лагерь и cGMP.[1][2]
Открытие
Существование Ca2+-стимулированная PDE1 была впервые продемонстрирована Cheung (1970), Kakiuchi и Yamazaki (1970) в результате их исследования бык мозг и крыса мозг соответственно.[1][3] С тех пор было обнаружено, что он широко распространен в различных млекопитающее ткани а также в других эукариоты. Сейчас это один из наиболее интенсивно изучаемых членов PDE суперсемейство ферментов,[3] что сегодня составляет 11 ген семьи,[1][4] и лучше всего охарактеризованный.[3]
Дальнейшие исследования в этой области наряду с повышением доступности моноклональные антитела показали, что различные PDE1 изоферменты существуют и были идентифицированы и очищены. Теперь известно, что PDE1 существует в виде тканеспецифичных изоферментов.[3]
Структура
Семейство изоферментов PDE1 относится к ферментам класса I,[2][5] который включает в себя все позвоночное животное PDE и некоторые дрожжи ферменты.[5] Все ферменты класса I имеют каталитическое ядро не менее 250 аминокислоты тогда как ферменты класса II не обладают такой общей чертой.[5]
Обычно ПДЭ позвоночных димеры линейной 50–150 кДа белки.[5] Они состоят из трех функциональных домены; консервативное каталитическое ядро, регуляторный N-конец и C-конец [3-5]. Белки химерный и каждый домен связан со своей конкретной функцией.[2]
Регуляторный N-конец существенно различается у разных типов PDE.[4][5] Они фланкируются каталитическим ядром и включают области, которые автоматически ингибируют каталитические домены. Они также нацелены на последовательности, которые контролируют субклеточный локализация. В PDE1 эта область содержит домен связывания кальмодулина.[4]
Каталитические домены PDE1 (и других типов PDE) имеют три спиральный субдомены: N-концевой участок складки циклина, линкерный участок и С-концевой спиральный пучок. Глубина гидрофобный карман формируется на интерфейсе этих подобластей. Он состоит из четырех подсайты. Они металл сайт связывания (M-сайт), сердцевинный карман (Q-карман), гидрофобный карман (H-карман) и область крышки (L-область). Сайт M помещается на дно гидрофобного кармана с несколькими металлическими атомы. Атомы металлов связываются с остатками, которые полностью консервативны у всех членов семейства PDE. Идентичность атомов металла неизвестна с полной уверенностью. Однако некоторые свидетельства указывают на то, что по крайней мере один из металлов цинк а другой, вероятно, будет магний. Цинк сфера координации состоит из трех гистидины, один аспартат и две воды молекулы. Координационная сфера магния включает тот же аспартат вместе с пятью молекулами воды, одна из которых является общей с молекулой цинка. Предполагаемая роль ионов металлов включает стабилизацию структуры, а также активацию гидроксид посредничать катализ.[4]
Домены разделены «шарнирными» участками, где их можно экспериментально разделить путем ограниченного протеолиза.[2]
Семейство изоферментов PDE1 (наряду с семейством PDE4) является наиболее разнообразным и включает многочисленные изоформы вариантов сплайсинга PDE1. Он имеет три подтипа, PDE1A, PDE1B и PDE1C, которые далее делятся на различные изоформы.[1][2]
Локализация
Локализация изоформ PDE1 в разных тканях / клетках и их расположение в клетках выглядит следующим образом:
| Изоформа | Тканевая / клеточная локализация | Внутриклеточная локализация |
|---|---|---|
| PDE1A (PDE1A ) | Гладкая мышца, сердце, легкие, мозг, сперма [2] | Преимущественно цитозольный [2] |
| PDE1A1 | Сердце, легкое [2] | Преимущественно цитозольный [2] |
| PDE1A2 | Мозг [2] | Преимущественно цитозольный [2] |
| PDE1B1 (PDE1B ) | Нейроны, лимфоциты, гладкие мышцы [2] мозг, сердце, скелетные мышцы [1] | Цитозольный [2] |
| PDE1B2 | Макрофаги, лимфоциты [2] | Цитозольный [2] |
| PDE1C (PDE1C ) | Мозг, разрастающиеся гладкие мышцы человека, сперматиды [2] | Цитозольный [2] |
| PDE1C1 | Мозг, сердце, яички [6] | - |
| PDE1C2 | Обонятельный эпителий [2] | Цитозольный [2] |
| PDE1C4 / 5 | мРНК присутствует в яичках [6] | - |
Таблица 1. Различное расположение PDE1 в тканях и внутри клеток.
Сообщается, что большинство изоформ PDE1 цитозольный. Однако есть случаи, когда PDE1 локализуются в субклеточных регионах, но мало что известно о молекулярном механизмы отвечает за такую локализацию. Считается вероятным, что уникальные N-концевые или C-концевые области различных изоформ позволяют нацеливать различные белки на конкретные субклеточные домены.[2]
Функциональная роль
PDE1 катализирует следующее химическая реакция:[7]
- Гидролитически удаляет 5'-нуклеотиды последовательно от 3'-гидрокси концы 3'-окси-концевых олигонуклеотиды
Гидролизует обе рибонуклеотиды и дезоксирибонуклеотиды. Имеет низкую активность по отношению к полинуклеотиды.
Внутриклеточные вторичные мессенджеры, такие как цГМФ и цАМФ, претерпевают быстрые изменения в концентрации в ответ на широкий спектр специфичных для клетки стимулов. Концентрация этих вторичных мессенджеров во многом определяется относительной синтетической активностью аденилатциклаза и деструктивная активность циклического нуклеотида PDE.[3] Роль ферментов PDE1 заключается в разложении цГМФ и цАМФ.[8]
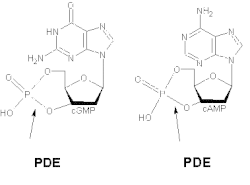
Различные изоформы проявляют разное сродство к цАМФ и цГМФ. PDE1A и PDE1B предпочтительно гидролизуют цГМФ, тогда как PDE1C разрушает цАМФ и цГМФ с высокой аффинностью. Например, в дыхательных путях гладкие мышцы у людей и других видов на общий PDE1 приходится более 50% гидролитической активности циклических нуклеотидов.[9] Было продемонстрировано, что удаление и чрезмерное выражение PDE1 оказывает сильное влияние на агонист -индуцирует передачу сигналов цАМФ, но мало влияет на базальный уровень цАМФ.[10]
Фармакология
Потому что in vitro регулирование Ca2+/ кальмодулин, считается, что PDE1 действуют как механизм интеграции клеточных сигнальных путей, опосредованных цГМФ и цАМФ, с путями, которые регулируют внутриклеточные уровни кальция.[2] Точная функция изоферментов PDE1 в различных патофизиологический процессы не ясны, потому что большинство исследований было проведено in vitro. Поэтому крайне важно направить дальнейшие исследования на исследования in vivo.[3]
Предполагается, что PDE1 играет роль в ряде физиологический и патологический процессы:
- PDE1A, скорее всего, служит для регулирования сосудистый концентрация гладкой мускулатуры, и было обнаружено, что она активируется в аорте крыс в ответ на хронические нитроглицерин лечение. Также возможно, что он играет роль в сперма функция.[8]
- PDE1B нокаутировать мыши увеличились локомотор активность и в некоторых парадигмы снижение памяти и обучаемости. PDE1B также участвует в дофаминергический сигнализация и индуцируется в нескольких типах активированных невосприимчивый клетки.[8] PDE1B мРНК индуцируется в PHA или активированных анти-CD3 / CD28 человеческих Т-лимфоциты и участвует в регуляции IL-13, участвующей в аллергический болезни.[1]
- Было показано, что PDE1C является основным регулятором пролиферации гладких мышц, по крайней мере, в гладких мышцах человека. Непролиферирующие гладкомышечные клетки (SMC) демонстрируют только низкие уровни PDE1C. выражение но он сильно выражен в пролиферирующих SMC. Следовательно, можно предположить, что торможение PDE1C может вызывать положительные эффекты из-за предполагаемого ингибирования пролиферации SMC, события, которое вносит важный вклад в патофизиологию атеросклероз.[8] Другая вероятная роль PDE1C заключается в обоняние [4], чтобы регулировать функцию сперматозоидов и нейронный сигнализация.[8]
Регулирование
Отличительной особенностью PDE1 как семейства является их регуляция кальцием (Ca2+) и кальмодулин (CaM).[11] Было показано, что кальмодулин активирует циклический нуклеотид PDE кальций-зависимым образом и кооперативное связывание четырех Ca2+ Кальмодулин необходим для полной активации ФДЭ1 [2]. Связывание одного Ca2+/ CaM комплекс на мономер к сайтам связывания около N-конца стимулирует гидролиз циклических нуклеотидов. В интактных клетках PDE1 почти исключительно активируется Ca2+ вход в камеру из внеклеточный Космос. Регулирование PDE1 с помощью Ca2+ и CaM был изучен in vitro, и эти исследования показали, что восемь метионин остатки внутри гидрофобных щелей Са2+-CaM необходимы для связывания и активации PDE1. Мутации в N-концевой доле CaM влияет на его способность активировать PDE1, поэтому считается, что C-концевая доля CaM служит для нацеливания CaM на PDE1, в то время как N-концевая доля активирует фермент. Наличие ароматный остаток, обычно триптофан, в СаМ-связывающей области Са2+-CaM-регулируемые белки также могут потребоваться для связывания с PDE1.[11]
Между разными изоферментами PDE1 существует значительная разница в близость для Ca2+/ CaM. Как правило, ферменты PDE1 обладают высоким сродством к комплексу, но на сродство может влиять фосфорилирование. Фосфорилирование PDE1A1 и PDE1A2 протеинкиназой A и PDE1B1 CaM Kinase II снижает их чувствительность к активации кальмодулина.[1] Это фосфорилирование может быть обращено фосфатазой кальциневрин.[2] Фосфорилирование изоферментов сопровождается снижением сродства изоферментов к СаМ, а также увеличением содержания Са2+ концентрации, необходимые для активации изоферментов СаМ.[3]
Ингибиторы и их функции
PDE проводились как терапевтический цели из-за основных фармакологический принцип, что регулирование деградации любого лиганд или же второй посланник может часто вызывать более быстрое и большее процентное изменение концентрации, чем сопоставимые темпы синтез. Другая причина заключается в том, что PDE не должны конкурировать с очень высокими уровнями эндогенный субстрата, чтобы быть эффективным, поскольку уровни цАМФ и цГМФ в большинстве клеток обычно находятся в микромолярный классифицировать.[2]
Доступность кристаллических структур с высоким разрешением каталитических доменов PDE делает разработку высокоэффективных и специфических ингибиторы возможный.[6]
Многие соединения, указанные как ингибиторы PDE1, не взаимодействуют напрямую с каталитическим сайтом PDE1, но взаимодействуют во время активации либо на уровне сайтов связывания кальмодулина, таких как соединение KS505a или прямо на Ca2+/ кальмодулин, такой как bepril, флунаризин и амиодарон.[1]
Те ингибиторы, которые взаимодействуют с каталитическим сайтом, занимают часть активного сайта, в основном вокруг Q-кармана и иногда вблизи M-кармана.[12] Основной точкой взаимодействия является консервативный гидрофобный карман, который участвует в ориентации пуринового кольца субстрата для взаимодействия с остатком глутамина, который имеет решающее значение для каталитического механизма PDE.[6]
В взаимодействия ингибиторов можно разделить на три основных типа: взаимодействия с ионами металлов, опосредованные водой, взаимодействия Н-связи с остатками белка, участвующие в распознавании нуклеотидов, и, что наиболее важно, взаимодействие с гидрофобными остатками, выстилающими полость активного центра. Похоже, что все известные ингибиторы используют эти три типа взаимодействий, и поэтому эти взаимодействия должны определять дизайн новых типов ингибиторов.[12]
Первоначально ингибиторы PDE1 были заявлены как эффективные сосудистые релаксанты. Однако, благодаря доступности очищенных клонированных ферментов, теперь известно, что такие ингибиторы фактически одинаково активны против PDE5.[4] Эти ингибиторы включают, например, запринаст, 8-метоксиметил IPMX и SCH 51866.[1]
Все терапевтически эффективные ингибиторы PDE должны быть включены в клетку, потому что все PDE локализованы в цитоплазме и / или на внутриклеточных мембранах.[8]
На сегодняшний день не существует реального и эффективного специфического ингибитора PDE1, который можно было бы использовать для оценки функциональной роли PDE1 в тканях.[1]
Общие ингибиторы


Нимодипин это дигидропиридин который противодействует / блокирует специфически L-тип Ca2+-канал, и впервые был описан как ингибитор PDE1. Этот эффект не связан с его свойствами антагониста кальция, поскольку он ингибирует в микромолярном диапазоне базальный и кальмодулин-стимулированный очищенный PDE1. Поскольку нимодипин в более низких концентрациях блокирует кальциевый канал L-типа, его можно использовать только для оценки участия PDE1 в тканевых и клеточных гомогенатах.[1]
Винпоцетин был описан как специфический ингибитор базальной и кальмодулин-активируемой PDE1. Этот эффект приводит к увеличению цАМФ по сравнению с цГМФ.[1][13] В основном он используется как фармакологический инструмент для выявления PDE1. Винпоцетин по-разному подавляет различные подтипы PDE1 (IC50 от 8 до 50 мкм), а также он способен ингибировать PDE7B. Его нельзя использовать в качестве специального инструмента для исследования функциональной роли PDE1 из-за его прямого активирующего воздействия на каналы BK (Ca).[1] Винпоцетин проникает через гематоэнцефалический барьер и поглощается тканями головного мозга. Было высказано предположение, что винпоцетин может влиять на потенциал-зависимые кальциевые каналы.[13]
IC224 ингибирует PDE1 (IC50 = 0,08 мкМ) с селективным отношением 127 (отношение IC50 значение для следующего наиболее чувствительного PDE и для IC50 значение для PDE1). Разработан корпорацией ICOS. Если IC224 аналогичным образом ингибирует базальный и активированный кальмодулином подтипы PDE1, это соединение может быть очень полезным для характеристики активности PDE1 и четкого исследования различных ролей PDE1 в патофизиологии.[1]
Ингибиторы при заболеваниях
Почти все фосфодиэстеразы экспрессируются в ЦНС, что делает это семейство генов привлекательным источником новых мишеней для лечения психиатрический и нейродегенеративный расстройства.[6]
PDE1A2 может играть потенциальную роль в нейродегенеративных заболеваниях, в том числе:[4]
- болезнь Паркинсона
- Аксональный нейрофиламент деградация
- Моторнейрональная деградация
- Ишемия нейронов
- Болезнь Альцгеймера
- Эпилепсия
PDE1C может играть роль в регуляции инсулин релиз [5] и может нацеливаться на пролиферирующие гладкомышечные клетки при атеросклеротических поражениях или во время рестеноз.[4][14]
Рекомендации
- ^ а б c d е ж грамм час я j k л м п Lugnier C (март 2006 г.). «Суперсемейство циклических нуклеотидфосфодиэстераз (PDE): новая мишень для разработки специфических терапевтических агентов». Pharmacol. Ther. 109 (3): 366–98. Дои:10.1016 / j.pharmthera.2005.07.003. PMID 16102838.
- ^ а б c d е ж грамм час я j k л м п о п q р s т ты v ш Бендер А.Т., Биво Д.А. (сентябрь 2006 г.). «Циклические нуклеотидные фосфодиэстеразы: молекулярная регуляция для клинического использования». Pharmacol. Rev. 58 (3): 488–520. Дои:10.1124 / пр.58.3.5. PMID 16968949.
- ^ а б c d е ж грамм Каккар Р., Раджу Р.В., Шарма Р.К. (июль 1999 г.). «Кальмодулин-зависимая циклическая нуклеотидфосфодиэстераза (PDE1)». Клетка. Мол. Life Sci. 55 (8–9): 1164–86. Дои:10.1007 / с000180050364. PMID 10442095.[постоянная мертвая ссылка ]
- ^ а б c d е ж грамм Jeon YH, Heo YS, Kim CM и др. (Июнь 2005 г.). «Фосфодиэстераза: обзор белковых структур, потенциальных терапевтических применений и последних достижений в разработке лекарств». Клетка. Мол. Life Sci. 62 (11): 1198–220. Дои:10.1007 / s00018-005-4533-5. PMID 15798894.
- ^ а б c d е ж Дуса Т.П. (январь 1999 г.). «Изоферменты циклической 3 ', 5'-нуклеотидной фосфодиэстеразы в клеточной биологии и патофизиологии почек». Почка Int. 55 (1): 29–62. Дои:10.1046 / j.1523-1755.1999.00233.x. PMID 9893113.
- ^ а б c d е Menniti FS, Faraci WS, Schmidt CJ (август 2006 г.). «Фосфодиэстеразы в ЦНС: мишени для разработки лекарств». Nat Rev Drug Discov. 5 (8): 660–70. Дои:10.1038 / nrd2058. PMID 16883304.
- ^ Хорана Г.Х. (1961). «Фосфодиэстеразы». В Boyer PD, Lardy H, Myrbäck K (ред.). Ферменты. 5 (2-е изд.). Нью-Йорк: Academic Press. С. 79–94.
- ^ а б c d е ж Бишофф Э (июнь 2004 г.). «Эффективность, селективность и последствия неселективности ингибирования ФДЭ». Int. J. Impot. Res. 16 (Приложение 1): S11–4. Дои:10.1038 / sj.ijir.3901208. PMID 15224129.
- ^ Гембиц М.А. (июнь 2005 г.). «Жизнь после PDE4: преодоление побочных эффектов с помощью ингибиторов фосфодиэстеразы двойной специфичности». Curr Opin Pharmacol. 5 (3): 238–44. Дои:10.1016 / j.coph.2005.04.001. PMID 15907909.
- ^ Thevelein JM, de Winde JH (сентябрь 1999 г.). «Новые механизмы восприятия и мишени для пути цАМФ-протеинкиназы A в дрожжах Saccharomyces cerevisiae». Мол. Микробиол. 33 (5): 904–18. Дои:10.1046 / j.1365-2958.1999.01538.x. PMID 10476026.
- ^ а б Горая Т.А., Купер Д.М. (июль 2005 г.). «Са2 + -кальмодулин-зависимая фосфодиэстераза (ФДЭ1): современные перспективы». Клетка. Сигнал. 17 (7): 789–97. Дои:10.1016 / j.cellsig.2004.12.017. PMID 15763421.
- ^ а б Card GL, England BP, Suzuki Y и др. (Декабрь 2004 г.). «Структурные основы действия препаратов, подавляющих фосфодиэстеразы». Структура. 12 (12): 2233–47. Дои:10.1016 / j.str.2004.10.004. PMID 15576036.
- ^ а б «Винпоцетин. Монография». Альтернативная медицина Rev. 7 (3): 240–3. Июнь 2002 г. PMID 12126465.
- ^ Мацумото Т., Кобаяси Т., Камата К. (август 2003 г.). «Фосфодиэстеразы в сосудистой системе». J Smooth Muscle Res. 39 (4): 67–86. Дои:10.1540 / jsmr.39.67. PMID 14692693.
внешняя ссылка
- Фосфодиэстераза + I в Национальной медицинской библиотеке США Рубрики медицинской тематики (MeSH)