Синтез жирных кислот - Википедия - Fatty acid synthesis
Синтез жирных кислот это создание жирные кислоты из ацетил-КоА и НАДФН через действие ферменты называется синтазы жирных кислот. Этот процесс происходит в цитоплазма из клетка. Большая часть ацетил-КоА, который превращается в жирные кислоты, происходит из углеводы через гликолитический путь. Гликолитический путь также обеспечивает глицерин с которым могут сочетаться три жирные кислоты (с помощью сложноэфирные связи ) сформировать триглицериды (также известные как «триацилглицерины» - чтобы отличить их от жирных «кислот» - или просто как «жир»), конечный продукт липогенный процесс. Когда только две жирные кислоты соединяются с глицерином и третья спиртовая группа фосфорилируется такой группой, как фосфатидилхолин, а фосфолипид сформирован. Фосфолипиды составляют основную часть липидные бислои которые составляют клеточные мембраны и окружить органеллы внутри ячеек (например, ядро клетки, митохондрии, эндоплазматический ретикулум, аппарат Гольджи так далее.)
Жирные кислоты с прямой цепью
Жирные кислоты с прямой цепью бывают двух типов: насыщенные и ненасыщенные.
Насыщенные жирные кислоты с прямой цепью

Так же, как β-окисление, синтез жирных кислот с прямой цепью происходит посредством шести повторяющихся реакций, показанных ниже, до тех пор, пока 16-углеродный пальмитиновая кислота производится.[1][2]
Представленные диаграммы показывают, как жирные кислоты синтезируются в микроорганизмах, и перечисляют ферменты, обнаруженные в кишечная палочка.[1] Эти реакции выполняются синтаза жирных кислот II (FASII), которые обычно содержат несколько ферментов, действующих как один комплекс. FASII присутствует в прокариоты, растения, грибы и паразиты, а также в митохондрии.[3]
У животных, а также у некоторых грибов, таких как дрожжи, эти же реакции происходят с синтазой жирных кислот I (FASI), большим димерным белком, который обладает всей ферментативной активностью, необходимой для образования жирной кислоты. FASI менее эффективен, чем FASII; тем не менее, он позволяет образовывать больше молекул, включая жирные кислоты со "средней длиной цепи", посредством обрыва цепи на ранней стадии.[3]
После образования жирной кислоты с 16: 0 углерода она может претерпевать ряд модификаций, приводящих к десатурации и / или удлинению. Элонгация, начиная со стеарата (18: 0), осуществляется в основном в ER несколькими мембраносвязанными ферментами. Ферментативные этапы, участвующие в процессе удлинения, в основном такие же, как и этапы, выполняемые FAS, но четыре основных последовательных этапа удлинения выполняются отдельными белками, которые могут быть физически связаны.[4][5]
| Шаг | Фермент | Реакция | Описание |
|---|---|---|---|
| (а) | Ацетил-КоА: ACP-трансацилаза | Активирует ацетил-КоА для реакции с малонил-ACP | |
| (б) | Малонил-КоА: АСР-трансацилаза | Активирует малонил-КоА для реакции с ацетил-ACP | |
| (c) | 3-кетоацил-ACP-синтаза | 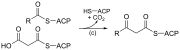 | Реагирует на ацильную цепь, связанную с ACP, с малонил-ACP, удлиняющим цепь. |
| (г) | 3-кетоацил-АСР редуктаза | Восстанавливает кетон углерода 3 до гидроксильной группы | |
| (е) | 3-гидроксиацил ACP дегидраза | Устраняет воду | |
| (е) | Эноил-АСР редуктаза | Восстанавливает двойную связь C2-C3. | |
| Сокращения: ACP - Белок-носитель ацила, CoA - Коэнзим А, НАДФ - Никотинамидадениндинуклеотидфосфат. | |||
Обратите внимание, что при синтезе жиров восстанавливающий агент НАДФН, в то время как НАД окислитель в бета-окисление (расщепление жирных кислот до ацетил-КоА). Это различие иллюстрирует общий принцип, согласно которому НАДФН расходуется во время биосинтетических реакций, тогда как НАДН генерируется в реакциях с выделением энергии.[6] (Таким образом, НАДФН также необходим для синтеза холестерин из ацетил-КоА; а НАДН образуется во время гликолиз.) Источник НАДФН двоякий. Когда малат окислительно декарбоксилируется "НАДФ"+-связанный яблочный фермент "с образованием пируват, CO2 и НАДФН образуются. НАДФН также образуется пентозофосфатный путь который превращает глюкозу в рибозу, которая может быть использована в синтезе нуклеотиды и нуклеиновые кислоты, или он может катаболизироваться до пирувата.[6]
Превращение углеводов в жирные кислоты
У человека жирные кислоты образуются из углеводов преимущественно в печень и жировая ткань, а также в молочные железы в период лактации.
Пируват, образующийся при гликолизе, является важным промежуточным звеном в превращении углеводов в жирные кислоты и холестерин.[6] Это происходит за счет превращения пирувата в ацетил-КоА в митохондриях. Однако этот ацетил-КоА необходимо транспортировать в цитозоль, где происходит синтез жирных кислот и холестерина. Это не может произойти напрямую. Для получения цитозольного ацетил-КоА цитрат (полученный конденсацией ацетил-КоА с оксалоацетатом) удаляется из цикл лимонной кислоты и переносится через внутреннюю митохондриальную мембрану в цитозоль.[6] Там он расщеплен Цитрат лиаза АТФ на ацетил-КоА и оксалоацетат. Оксалоацетат можно использовать для глюконеогенез (в печени), или он может быть возвращен в митохондрии в виде малата.[7] Цитозольный ацетил-КоА карбоксилируется ацетил-КоА карбоксилаза в малонил-КоА, первый совершенный шаг в синтезе жирных кислот.[7][8]
Животные не могут повторно синтезировать углеводы из жирных кислот
Основное топливо, которое хранится в организме животных, - жир. Жировые запасы молодого взрослого человека составляют в среднем около 15-20 кг, но сильно варьируются в зависимости от возраста, пола и индивидуального характера.[9] Напротив, человеческое тело хранит только около 400 г гликоген, из которых 300 г заблокированы внутри скелетных мышц и недоступны для организма в целом. Примерно 100 г гликогена, хранящегося в печени, истощаются в течение одного дня голодания.[10] После этого глюкоза, которая выделяется в кровь печенью для общего использования тканями организма, должна быть синтезирована из глюкогенные аминокислоты и еще несколько глюконеогенные субстраты, в состав которых не входят жирные кислоты.[11]
Жирные кислоты расщепляются до ацетил-КоА с помощью бета-окисление внутри митохондрий, тогда как жирные кислоты синтезируются из ацетил-КоА вне митохондрии, в цитозоле. Эти два пути различны не только в том, где они происходят, но также в протекающих реакциях и используемых субстратах. Эти два пути являются взаимно ингибирующими, предотвращая попадание ацетил-КоА, продуцируемого бета-окислением, в синтетический путь через ацетил-КоА карбоксилаза реакция.[11] Его также нельзя преобразовать в пируват как декарбоксилирование пирувата реакция необратима.[10] Вместо этого он конденсируется с оксалоацетат, чтобы войти в цикл лимонной кислоты. Во время каждого витка цикла два атома углерода покидают цикл как CO2 в реакциях декарбоксилирования, катализируемых изоцитратдегидрогеназа и альфа-кетоглутаратдегидрогеназа. Таким образом, каждый поворот цикла лимонной кислоты окисляет звено ацетил-КоА, одновременно регенерируя молекулу оксалоацетата, с которой ацетил-КоА первоначально объединился с образованием лимонная кислота. Реакции декарбоксилирования происходят до малат образуется в цикле. Это единственное вещество, которое можно удалить из митохондрии и попасть в глюконеогенный путь с образованием глюкозы или гликогена в печени или любой другой ткани.[11] Следовательно, не может быть чистого преобразования жирных кислот в глюкозу.
Только растения обладают ферментами, превращающими ацетил-КоА в оксалоацетат, из которого может быть образован малат, который в конечном итоге превратится в глюкозу.[11]
Регулирование
Ацетил-КоА образуется в малонил-КоА посредством ацетил-КоА карбоксилаза, в этот момент малонил-КоА предназначен для включения в путь синтеза жирных кислот. Ацетил-КоА-карбоксилаза является точкой регуляции в синтезе насыщенных жирных кислот с прямой цепью и подвержена обоим фосфорилирование и аллостерическая регуляция. Регуляция фосфорилированием происходит в основном у млекопитающих, тогда как аллостерическая регуляция происходит у большинства организмов. Аллостерический контроль происходит в виде ингибирования по обратной связи пальмитоил-КоА и активации цитратом. Когда имеется высокий уровень пальмитоил-КоА, конечного продукта синтеза насыщенных жирных кислот, он аллостерически инактивирует ацетил-КоА-карбоксилазу, чтобы предотвратить накопление жирных кислот в клетках. Цитрат активирует ацетил-КоА карбоксилазу при высоких уровнях, потому что высокие уровни указывают на то, что ацетил-КоА достаточно для питания Цикл Кребса и сберечь энергию.[12]
Высокие уровни в плазме инсулин в плазме крови (например, после еды) вызывают дефосфорилирование ацетил-КоА-карбоксилазы, тем самым способствуя образованию малонил-КоА из ацетил-КоА и, следовательно, превращению углеводов в жирные кислоты, в то время как адреналин и глюкагон (попадает в кровь во время голодания и физических упражнений) вызывают фосфорилирование этого фермента, подавляя липогенез в пользу окисления жирных кислот через бета-окисление.[6][8]
Ненасыщенные жирные кислоты с прямой цепью
Анаэробная десатурация
Многие бактерии используют анаэробный путь для синтеза ненасыщенных жирных кислот. Этот путь не использует кислород и зависит от ферментов, которые вставляют двойную связь перед удлинением, используя нормальный механизм синтеза жирных кислот. В кишечная палочкаэтот путь хорошо изучен.

- FabA представляет собой β-гидроксидеканоил-ACP-дегидразу - он специфичен для промежуточного соединения синтеза 10-углеродных насыщенных жирных кислот (β-гидроксидеканоил-ACP).
- FabA катализирует дегидратацию β-гидроксидеканоил-ACP, вызывая высвобождение воды и вставку двойной связи между C7 и C8, считая от метильного конца. Это создает промежуточный транс-2-деценоил.
- Либо транс-2-деценоил-промежуточный продукт может быть переведен на нормальный путь синтеза насыщенных жирных кислот с помощью FabB, где двойная связь будет гидролизована и конечный продукт будет насыщенной жирной кислотой, либо FabA будет катализировать изомеризацию в цис- 3-деценоил промежуточный.
- FabB представляет собой β-кетоацил-АСР-синтазу, которая удлиняет и направляет промежуточные соединения в основной путь синтеза жирных кислот. Когда FabB реагирует с промежуточным цис-деценоилом, конечный продукт после удлинения будет ненасыщенной жирной кислотой.[13]
- Две основные получаемые ненасыщенные жирные кислоты - это пальмитолеил-ACP (16: 1ω7) и цис-вакценоил-ACP (18: 1ω7).[14]
Большинство бактерий, подвергающихся анаэробной десатурации, содержат гомологи FabA и FabB.[15] Клостридии - главное исключение; у них есть новый фермент, который еще предстоит идентифицировать, который катализирует образование двойной цис-связи.[14]
Регулирование
Этот путь проходит транскрипционная регуляция к FadR и FabR. FadR является более изученным белком, которому приписывают бифункциональные характеристики. Он действует как активатор FabA и fabB транскрипция и как репрессор для β-окисления регулон. Напротив, FabR действует как репрессор транскрипции fabA и fabB.[13]
Аэробная десатурация
Аэробная десатурация - наиболее распространенный путь синтеза ненасыщенных жирных кислот. Он используется у всех эукариот и некоторых прокариот. Этот путь использует десатуразы для синтеза ненасыщенных жирных кислот из полноразмерных субстратов насыщенных жирных кислот.[16] Все десатуразы требуют кислорода и в конечном итоге потребляют НАДН, даже если десатурация является окислительным процессом. Десатуразы специфичны для двойной связи, которую они индуцируют в субстрате. В Bacillus subtilis, десатураза Δ5-Des, специфичен для индукции цис-двойной связи при Δ5 позиция.[7][16] Saccharomyces cerevisiae содержит одну десатуразу, Ole1p, которая индуцирует цис-двойную связь при Δ9.[7]
У млекопитающих аэробная десатурация катализируется комплексом из трех мембраносвязанных ферментов (НАДН-цитохром b5 редуктаза, цитохром b5, а десатураза). Эти ферменты пропускают молекулярный кислород, O2, чтобы взаимодействовать с насыщенной жирной цепью ацил-КоА, образуя двойную связь и две молекулы воды, H2О. Два электрона происходят от НАДН + Н+ и два от одинарной связи в цепи жирной кислоты.[6] Однако эти ферменты млекопитающих неспособны создавать двойные связи у атомов углерода за пределами С-9 в цепи жирной кислоты.[nb 1].) Следовательно, млекопитающие не могут синтезировать линолеат или же линоленат (которые имеют двойные связи в C-12 (= Δ12), или C-12 и C-15 (= Δ12 и Δ15) положениях соответственно, а также на Δ9 положение), ни полиненасыщенный, 20-углеродный арахидоновая кислота который получен из линолеата. Все это называется незаменимые жирные кислоты Это означает, что они необходимы организму, но могут поступать только с пищей. (Арахидоновая кислота является предшественником простагландины которые выполняют широкий спектр функций как местные гормоны.)[6]
Жирные кислоты с нечетной цепью
Жирные кислоты с нечетной цепью (OCFA) - это те жирные кислоты которые содержат нечетное количество атомов углерода. Наиболее распространенными OCFA являются насыщенные производные C15 и C17 соответственно. пентадекановая кислота и гептадекановая кислота.[17] Синтез четных цепочек жирная кислота синтез осуществляется путем сборки ацетил-КоА предшественники, однако, пропионил-КоА вместо ацетил-КоА используется в качестве праймера для биосинтеза длинноцепочечных жирных кислот с нечетным числом атомов углерода.[18]
РегулированиеВ Б. subtilis, этот путь регулируется двухкомпонентная система: DesK и DesR. DesK представляет собой мембранно-ассоциированную киназу, а DesR - транскрипционный регулятор des ген.[7][16] Регулировка реагирует на температуру; когда происходит падение температуры, этот ген активируется. Ненасыщенные жирные кислоты увеличивают текучесть мембраны и стабилизируют ее при более низких температурах. DesK - это сенсорный белок, который при понижении температуры будет аутофосфорилироваться. DesK-P будет передавать свою фосфорильную группу DesR. Два белка DesR-P будут димеризоваться и связываться с промоторами ДНК des ген и рекрутировать РНК-полимеразу для начала транскрипции.[7][16]
Синегнойная палочка
Как правило, анаэробный и аэробный синтез ненасыщенных жирных кислот не происходит в одной и той же системе, однако Синегнойная палочка и Вибрион ABE-1 - исключения.[19][20][21]Пока P. aeruginosa претерпевает в основном анаэробную десатурацию, а также проходит два аэробных пути. Один путь использует Δ9-десатураза (DesA), которая катализирует образование двойной связи в липидах мембран. Другой путь использует два белка, DesC и DesB, вместе, чтобы действовать как Δ9-десатураза, которая вставляет двойную связь в молекулу насыщенной жирной кислоты-КоА. Этот второй путь регулируется репрессорным белком DesT. DesT также подавляет FabAB выражение для анаэробной десатурации в присутствии экзогенных ненасыщенных жирных кислот. Это функция для координации экспрессии двух путей в организме.[20][22]
Жирные кислоты с разветвленной цепью
Жирные кислоты с разветвленной цепью обычно являются насыщенными и относятся к двум различным семействам: изо-серии и антеизо-серии. Было обнаружено, что Актиномицеты содержат уникальные механизмы синтеза жирных кислот с разветвленной цепью, в том числе те, которые образуют туберкулостериновую кислоту.
Система синтеза жирных кислот с разветвленной цепью



Система синтеза жирных кислот с разветвленной цепью использует α-кетокислоты в качестве грунтовки. Эта система отличается от синтетазы жирных кислот с разветвленной цепью, которая использует эфиры ацил-КоА с короткой цепью в качестве праймеров.[23] праймеры α-кетокислот происходят из трансаминирование и декарбоксилирование из валин, лейцин, и изолейцин с образованием 2-метилпропанил-КоА, 3-метилбутирил-КоА и 2-метилбутирил-КоА соответственно.[24] Праймеры 2-метилпропанил-КоА, полученные из валина, имеют удлиненную форму для образования жирных кислот изо-ряда с четным номером, таких как 14-метилпентадекановая (изопальмитиновая) кислота, а праймеры 3-метилбутирил-КоА из лейцина могут использоваться для образования нечетных жирных кислот. жирные кислоты изо-ряда, такие как 13-метилтетрадекановая кислота. Праймеры 2-метилбутирил-КоА из изолейцина имеют удлиненную форму с образованием жирных кислот антеизо-ряда, содержащих нечетное число атомов углерода, таких как 12-метилтетрадекановая кислота.[25] Декарбоксилирование предшественников праймеров происходит через декарбоксилаза α-кетокислот с разветвленной цепью (BCKA) фермент. Удлинение жирной кислоты происходит по тому же пути биосинтеза в кишечная палочка используется для производства жирных кислот с прямой цепью, где малонил-КоА используется в качестве удлинителя цепи.[26] Основными конечными продуктами являются жирные кислоты с разветвленной цепью, состоящие из 12–17 атомов углерода, и их состав обычно является однородным и характерным для многих видов бактерий.[25]
Декарбоксилаза BCKA и относительная активность субстратов α-кетокислот
Фермент декарбоксилазы BCKA состоит из двух субъединиц в тетрамерной структуре (A2B2) и необходим для синтеза жирных кислот с разветвленной цепью. Он отвечает за декарбоксилирование α-кетокислот, образованных переаминированием валина, лейцина и изолейцина, и производит праймеры, используемые для синтеза жирных кислот с разветвленной цепью. Активность этого фермента намного выше с субстратами α-кетокислот с разветвленной цепью, чем с субстратами с прямой цепью, а в Бациллы его специфичность наиболее высока для производной изолейцина α-кето-β-метилвалериановой кислоты, за которой следует α-кетоизокапроат и α-кетоизовалерат.[25][26] Высокое сродство фермента к α-кетокислотам с разветвленной цепью позволяет ему функционировать как система, дающая праймеры для синтетазы жирных кислот с разветвленной цепью.[26]
| Субстрат | Деятельность BCKA | Произведенный CO2 (нмоль / мин, мг) | Км (мкМ) | Vmax (нмоль / мин, мг) |
|---|---|---|---|---|
| L-α-кето-β-метилвалерат | 100% | 19.7 | <1 | 17.8 |
| α-кетоизовалерат | 63% | 12.4 | <1 | 13.3 |
| α-кетоизокапроат | 38% | 7.4 | <1 | 5.6 |
| Пируват | 25% | 4.9 | 51.1 | 15.2 |
Факторы, влияющие на длину цепи и распределение рисунка
Праймеры α-кетокислот используются для получения жирных кислот с разветвленной цепью, которые, как правило, имеют длину от 12 до 17 атомов углерода. Пропорции этих жирных кислот с разветвленной цепью имеют тенденцию быть однородными и согласованными среди конкретных видов бактерий, но могут быть изменены из-за изменений в концентрации малонил-КоА, температуры или присутствующих факторов термостабильности (HSF).[25] Все эти факторы могут влиять на длину цепи, и было продемонстрировано, что HSF изменяют специфичность декарбоксилазы BCKA к определенному субстрату α-кетокислот, таким образом изменяя соотношение продуцируемых жирных кислот с разветвленной цепью.[25] Было показано, что увеличение концентрации малонил-КоА приводит к увеличению продуцирования жирных кислот C17 до тех пор, пока не будет достигнута оптимальная концентрация (≈20 мкм) малонил-КоА. Понижение температуры также имеет тенденцию немного сдвигать распределение жирных кислот в сторону жирных кислот C17 в Бациллы разновидность.[23][25]
Синтаза жирных кислот с разветвленной цепью
Эта система функционирует аналогично системе синтеза жирных кислот с разветвленной цепью, однако в ней используются карбоновые кислоты с короткой цепью в качестве праймеров вместо альфа-кетокислот. Как правило, этот метод используется бактериями, которые не способны выполнять систему жирных кислот с разветвленной цепью с использованием альфа-кето праймеров. Типичные короткоцепочечные праймеры включают изовалерат, изобутират и 2-метилбутират. Как правило, кислоты, необходимые для этих грунтовок, поступают из окружающей среды; это часто наблюдается у бактерий рубца.[27]
Общая реакция:
- Изобутирил-КоА + 6 малонил-КоА + 12 НАДФН + 12Н+ → Изопальмитиновая кислота + 6 CO2 12 НАДФ + 5 ч2O + 7 CoA[23]
Разница между (линейной) синтазой жирных кислот и синтазой жирных кислот с разветвленной цепью заключается в субстратной специфичности фермента, который катализирует реакцию ацил-КоА с ацил-АСР.[23]
Омега-алициклические жирные кислоты
Омега-алициклические жирные кислоты обычно содержат омега-концевую пропильную или бутирильную циклическую группу и являются одними из основных мембранных жирных кислот, обнаруживаемых у нескольких видов бактерий. Синтетаза жирных кислот, используемая для производства омега-алициклических жирных кислот, также используется для получения мембранных жирных кислот с разветвленной цепью. У бактерий с мембранами, состоящими в основном из омега-алициклических жирных кислот, количество сложных эфиров циклических карбоновых кислот и CoA намного больше, чем у праймеров с разветвленной цепью.[23] Синтез циклических праймеров не совсем понятен, но было высказано предположение, что механизм включает превращение сахаров в шикимовая кислота который затем превращается в эфиры циклогексилкарбоновой кислоты-CoA, которые служат праймерами для синтеза омега-алициклических жирных кислот[27]
Синтез туберкулостеариновой кислоты

Туберкулостеариновая кислота (D-10-метилстеариновая кислота) представляет собой насыщенную жирную кислоту, которая, как известно, производится Микобактерии виды и два вида Streptomyces. Он образуется из предшественника олеиновой кислоты (мононенасыщенной жирной кислоты).[28] После этерификации олеиновой кислоты до фосфолипида S-аденозилметионин отдает метильную группу двойной связи олеиновой кислоты.[29] Эта реакция метилирования образует промежуточное соединение 10-метилен-октадеканоал. Последовательное восстановление остатка с НАДФН в качестве кофактора приводит к 10-метилстеариновой кислоте.[24]
Смотрите также
Примечание ноги
- ^ Положение атомов углерода в жирной кислоте может быть обозначен с конца COOH- (или карбокси), или с -CH3 (или метиловый) конец. Если указано с конца -COOH, то используются обозначения C-1, C-2, C-3, .... (И т. Д.) (Синие цифры на диаграмме справа, где C-1 - это - Углерод COOH). Если позиция отсчитывается от другой, -CH3, конец, то позиция обозначается обозначением ω-n (цифры красного цвета, где ω-1 относится к метильному углероду).
 Нумерация атомов углерода
Нумерация атомов углеродаТаким образом, положения двойных связей в цепи жирных кислот можно указывать двумя способами, используя обозначение C-n или ω-n. Таким образом, в жирной кислоте из 18 атомов углерода двойная связь между C-12 (или ω-7) и C-13 (или ω-6) обозначается как Δ12 если отсчитывать от конца -COOH (указывающего только «начало» двойной связи), или как ω-6 (или омега-6), если отсчитывать от -CH3 конец. «Δ» - это греческая буква «дельта», которая переводится как «D» (т.е. Dдвойная связь) в латинском алфавите. Омега (ω) - последняя буква в греческом алфавите, и поэтому используется для обозначения «последнего» атома углерода в цепи жирной кислоты. Поскольку обозначение ω-n используется почти исключительно для обозначения положений двойных связей, близких к -CH3 закончиться незаменимые жирные кислоты, нет необходимости в эквивалентном "Δ" -подобном обозначении - использование обозначения "ω-n" всегда относится к положению двойной связи.

Рекомендации
- ^ а б Дейкстра, Альберт Дж., Р. Дж. Гамильтон и Вольф Хэмм. «Биосинтез жирных кислот». Транс-жирные кислоты. Оксфорд: Blackwell Pub., 2008. 12. Печать.
- ^ «Путь MetaCyc: суперпуть биосинтеза жирных кислот (Кишечная палочка)".
- ^ а б «Жирные кислоты: насыщенные с прямой цепью, структура, возникновение и биосинтез». Липидная библиотека - химия липидов, биология, технология и анализ. Интернет. 30 апреля 2011 г. <http://lipidlibrary.aocs.org/lipids/fa_sat/index.htm В архиве 21 июля 2011 г. Wayback Machine >.
- ^ «Путь MetaCyc: биосинтез стеарата I (животные)».
- ^ «Путь MetaCyc: биосинтез очень длинноцепочечных жирных кислот II».
- ^ а б c d е ж грамм Страйер, Люберт (1995). Биохимия (Четвертое изд.). Нью-Йорк: W.H. Фримен и компания. С. 559–565, 614–623. ISBN 0-7167-2009-4.
- ^ а б c d е ж Ferre, P .; Ф. Фуфель (2007). «Фактор транскрипции SREBP-1c и липидный гомеостаз: клиническая перспектива». Гормональные исследования. 68 (2): 72–82. Дои:10.1159/000100426. PMID 17344645. Получено 30 августа 2010.
этот процесс показан графически на странице 73
- ^ а б Воет, Дональд; Джудит Г. Воет; Шарлотта В. Пратт (2006). Основы биохимии, 2-е издание. John Wiley and Sons, Inc., стр.547, 556. ISBN 0-471-21495-7.
- ^ Sloan, A.W; Koeslag, J.H .; Бределл, Г.А.Г. (1973). «Работоспособность и работоспособность по составу тела юношей активного и малоактивного». Европейский журнал прикладной физиологии. 32: 17–24. Дои:10.1007 / bf00422426. S2CID 39812342.
- ^ а б Страйер, Люберт (1995). Биохимия (Четвертое изд.). Нью-Йорк: W.H. Фримен и компания. С. 581–602, 613, 775–778. ISBN 0-7167-2009-4.
- ^ а б c d Страйер, Люберт (1995). «Метаболизм жирных кислот». В: Биохимия. (Четвертое изд.). Нью-Йорк: W.H. Фримен и компания. С. 603–628. ISBN 0-7167-2009-4.
- ^ Диван, Джойс Дж. «Синтез жирных кислот». Политехнический институт Ренсселера (RPI) :: Архитектура, бизнес, инженерия, информационные технологии, гуманитарные науки, наука. Интернет. 30 апреля 2011 г. <http://rpi.edu/dept/bcbp/molbiochem/MBWeb/mb2/part1/fasynthesis.htm В архиве 7 июня 2011 г. Wayback Machine >.
- ^ а б Фэн, Юджун и Джон Эронан. «Комплексное связывание репрессора FabR бактериального биосинтеза ненасыщенных жирных кислот с его родственными промоторами». Молекулярная микробиология 80.1 (2011): 195–218.
- ^ а б Чжу, Лей и др. «Функции белков FabF и FabZ Clostridium acetobutylicium в биосинтезе ненасыщенных жирных кислот». BMC Microbiology 9 (2009): 119.
- ^ Ван, Хайхонг и Джон Э.Кронан. «Функциональная замена белков FabA и FabB в синтезе жирных кислот Escherichia coli на гомологи FabZ и FabF Enterococcus faecalis». Журнал биологической химии 279.33 (2004): 34489-95.
- ^ а б c d Мансилла, Мара С. и Диего Мендоса. «Bacillus subtilis desaturase: модель для понимания модификации фосфолипидов и измерения температуры». Архив микробиологии 183.4 (2005): 229-35.
- ^ Пфеффер, Мария; Jaudszus, Анке (2016). «Пентадекановая и гептадекановая кислоты: многогранные жирные кислоты с нечетной цепью». Достижения в области питания: международный обзорный журнал. 7 (4): 730–734. Дои:10.3945 / an.115.011387. ЧВК 4942867. PMID 27422507.
- ^ Смит, С. (1994). «Синтаза жирных кислот животных: один ген, один полипептид, семь ферментов». Журнал FASEB. 8 (15): 1248–1259. Дои:10.1096 / fasebj.8.15.8001737. PMID 8001737. S2CID 22853095.
- ^ Вада, М., Н. Фукунага и С. Сасаки. «Механизм биосинтеза ненасыщенных жирных кислот в штамме Pseudomonas sp. E-3, психротрофной бактерии». Журнал бактериологии 171.8 (1989): 4267-71.
- ^ а б Субраманиан, Читра, Чарльз Орук и Юн-Мейчжан. «DesT координирует экспрессию анаэробных и аэробных путей биосинтеза ненасыщенных жирных кислот у Pseudomonas aeruginosa». Журнал бактериологии 192.1 (2010): 280-5.
- ^ Morita, N, et al. «Как анаэробный путь, так и аэробная десатурация участвуют в синтезе ненасыщенных жирных кислот в штамме Vibrio sp. ABE-1». Письма FEBS 297.1–2 (1992): 9–12.
- ^ Чжу, Кун и др. «Два аэробных пути образования ненасыщенных жирных кислот у Pseudomonas aeruginosa». Молекулярная микробиология 60.2 (2006): 260-73.
- ^ а б c d е Канеда, Тоши. «Изо- и антеизо-жирные кислоты в бактериях: биосинтез, функция и таксономическое значение». Microbiological Reviews 55.2 (1991): 288–302.
- ^ а б «Жирные кислоты с разветвленной цепью, фитановая кислота, туберкулостеариновая кислота, изо / антеизо-жирные кислоты». Липидная библиотека - химия липидов, биология, технология и анализ. Интернет. 1 мая 2011 г. «Архивная копия». Архивировано из оригинал 12 января 2010 г.. Получено 8 марта 2014.CS1 maint: заархивированная копия как заголовок (связь).
- ^ а б c d е ж Наик, Деварай Н. и Тоши Канеда. «Биосинтез разветвленных длинноцепочечных жирных кислот видами Bacillus: относительная активность трех α-кетокислотных субстратов и факторов, влияющих на длину цепи». Может. J. Microbiol. 20 (1974): 1701–708.
- ^ а б c Оку, Хиросуке и Тоши Канеда. «Биосинтез жирных кислот с разветвленной цепью в Bacillis Subtilis». Журнал биологической химии 263.34 (1988): 18386-8396.
- ^ а б Кристи, Уильям В. "Жирные кислоты: природные алициклические структуры, возникновение и биохимия". Липидная библиотека AOCS. 5 апреля 2011 г. Интернет. 24 апреля 2011 г. <«Архивная копия» (PDF). Архивировано из оригинал (PDF) 21 июля 2011 г.. Получено 2 мая 2011.CS1 maint: заархивированная копия как заголовок (связь)>.
- ^ Рэтледж, Колин и Джон Стэнфорд. Биология микобактерий. Лондон: Академический, 1982. Печать.
- ^ Кубица, Джордж П. и Лоуренс Г. Уэйн. Микобактерии: Справочник. Нью-Йорк: Деккер, 1984. Печать.
внешняя ссылка
Метаболизм карта | ||
|---|---|---|
Одиночные линии: пути, общие для большинства форм жизни. Двойные линии: пути не у человека (встречаются, например, у растений, грибов, прокариот). | ||
